Advertisement
Grab your lab coat. Let's get started
Welcome!
Welcome!
Create an account below to get 6 C&EN articles per month, receive newsletters and more - all free.
It seems this is your first time logging in online. Please enter the following information to continue.
As an ACS member you automatically get access to this site. All we need is few more details to create your reading experience.
Not you? Sign in with a different account.
Not you? Sign in with a different account.
ERROR 1
ERROR 1
ERROR 2
ERROR 2
ERROR 2
ERROR 2
ERROR 2
Password and Confirm password must match.
If you have an ACS member number, please enter it here so we can link this account to your membership. (optional)
ERROR 2
ACS values your privacy. By submitting your information, you are gaining access to C&EN and subscribing to our weekly newsletter. We use the information you provide to make your reading experience better, and we will never sell your data to third party members.
Analytical Chemistry
DNA Sequencing Forges Ahead
Next-generation technologies lower cost of genome sequencing
by Celia Henry Arnaud
December 14, 2009
| A version of this story appeared in
Volume 87, Issue 50
The first human genome sequence cost more than $2 billion and took about a decade to complete. Achieving the milestone of the billionth base sequenced in the 3 billion-base-pair human genome was a cause for celebration.
Since the end of the Human Genome Project in 2003, cost and time barriers in sequencing methods have been shattered multiple times, and they keep getting lower. Technology has now advanced to the point that sequencing a genome can take less than a month and with some platforms, less than a week. Sequencing a billion bases is now the work of a single day. And the cost? Depending on the platform and whom you ask, reagents to sequence a billion bases now run as little as $5,000, although instrumentation, facility, and personnel costs must be added to that.
The technological improvements and plummeting costs of sequencing are paving the way for whole-genome sequencing for research purposes and, ultimately, affordable sequencing of personal genomes. Scientists interested in genome sequencing can now choose from a number of sequencing technologies that ease sample preparation and allow longer stretches of DNA to be read at a time. And there are more technologies to come.
The Human Genome Project obtained the first human sequence with the Sanger method of DNA sequencing. This laborious method involves copying a DNA template in the presence of dye-labeled modified nucleotides that terminate DNA-strand elongation when they are incorporated. Because these modified nucleotides can be incorporated at any point in the strands, the sequencing reaction results in a mixture of DNA strands of different length, each with its final base labeled with a fluorescent dye. These strands are separated by size with capillary electrophoresis. Lining the strands up in order creates a ladder that allows the sequence to be read easily. Although the Sanger method remains the gold standard for DNA sequencing in terms of accuracy, it is being supplanted by more efficient methods.
Many of the so-called second-generation sequencing methods use an approach in which DNA is sequenced by replicating a template and noting the identity of each base after its incorporation into the growing strand. Most commercially available technologies can accurately “read” a relatively small number of bases—generally up to 35 to 100—at one time.
Just like Sanger sequencing, most current methods require extensive sample preparation that involves making and amplifying a library of fragments from genomic DNA. But unlike Sanger sequencing, no lengthy separation step is required, and the methods are massively parallel. A genome sequence is assembled by aligning millions of these fragments against the reference sequence from the Human Genome Project. Because of the short read lengths and because of potential errors in base identification, each base must be identified many times. The higher this “fold coverage”—often about 30×—the higher the confidence in putative deviations, such as single-nucleotide polymorphisms, from the reference sequence.
Generally, bases are added one at a time and allowed to react. Excess reagent is rinsed out, and the device is then imaged to see where a base was incorporated. When a base is incorporated into a growing DNA strand, it gives off a detectable signal. The nature of that signal is the primary difference between versions of the method.
The first of these methods for whole-genome sequencing was commercialized by 454 Life Sciences, in Branford, Conn. In that company’s version, called pyrosequencing, the nucleotides release a flash of chemiluminescent light when they are incorporated into the growing strand. 454’s technology achieves longer read lengths than other available methods, currently about 500 bases, according to Michael Egholm, vice president of research and development. “We’re working on doubling the read length,” he says.
Another pioneer of second-generation DNA sequencing is San Diego-based Illumina. In Illumina’s technology, synthesis occurs on an array of randomly addressable clusters, each one with a different portion of the DNA sequence attached to it. All four bases can be added simultaneously because each base has a different fluorescent label. This reduces the number of cycles needed for a given read length and speeds up the run time.
Illumina has increased its read lengths from about 35 bases to 100 bases while reducing the time required for each cycle. “The time the instrument runs is the product of the number of cycles and the time for each cycle,” says Jay T. Flatley, president and chief executive officer at Illumina. “As our read lengths have gone up, we’ve reduced the cycle time enough that the overall run time hasn’t gone up very much.”

Helicos Biosciences, in Cambridge, Mass., also uses sequencing by synthesis, but the firm sequences single molecules rather than making many copies.
“We’re the only company that actually sequences the molecules of DNA that come out of your cell” instead of cloned and amplified DNA, says Patrice Milos, chief scientific officer at Helicos. “Because we’re sequencing the DNA molecules that come from your cells, there are no libraries created, there’s no ligation, there’s no sample amplification.” The sample preparation is therefore easier, and potential sources of bias are eliminated.
DNA extracted from the cells is broken into pieces, and the end of each fragment is tagged with a “polyA tail,” a string of adenosine nucleotides. The polyA tail allows the DNA to be attached to the surface of a flow cell. The technology now requires the amount of DNA in 100 to 200 cells to sequence an entire genome, but the company aspires to single-cell analysis, Milos says.
Pacific Biosciences’ technology puts a different twist on single-molecule sequencing by synthesis. Most of these methods treat the polymerase enzyme as just another reagent to be added and rinsed away with each cycle of base addition.
“Our view is that this enzyme is really a sequencing instrument in and of itself, and what a horrible shame to throw it away after every base you sequence,” says Stephen Turner, chief technology officer at PacBio, in Menlo Park, Calif. “If we free it up to do what nature has programmed it over billions of years of evolution to do, we can get the extraordinary features that it has of extreme frugality and high speed.”
A crucial component of PacBio’s technology is that fluorescent dyes are attached to the terminal phosphate of each nucleotide instead of the base, Turner says. Attaching the dye to the phosphate means that it will be spontaneously removed as part of the nucleotide incorporation, eliminating the need for a separate step to cleave off the dye.
PacBio’s sequencing chip is made of an array of zero-mode waveguides. Each waveguide is a hole tens of nanometers in diameter fabricated in a metal film on a silicon dioxide substrate. The 20-zeptoliter detection volume is small enough to detect the fluorescence from each incorporated nucleotide against a background of all the other labeled nucleotides floating in solution. When the polymerase cleaves off the label, the dye diffuses out of the detection volume.
The firm’s technology enables long read lengths of thousands of bases. The read length is limited by the dye photochemistry, which can lead to damage that shuts down the enzyme, and by the processivity of the polymerase, or how long it remains associated with the template strand. The enzyme can incorporate about 100,000 bases before it dissociates from the template, although PacBio plans to start with much shorter read lengths of up to a few thousand bases. PacBio is set to release its instrument in the second half of 2010, Turner says.
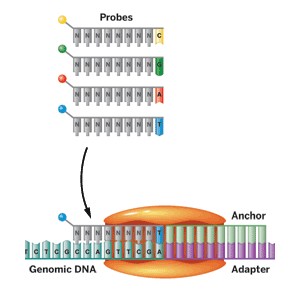
Other companies, such as Applied Biosystems (a division of Life Technologies), Dover-Polonator (a division of Danaher), and Complete Genomics, sequence DNA using variants of a method called sequencing by ligation. In Complete Genomics’ method, fluorescently labeled probe molecules are used to read seven to 10 bases adjacent to an adapter site inserted into a DNA sample. An anchor hybridizes to the adapter site, and the probe molecules compete for ligation to that anchor. A probe succeeds only if it is complementary to the sequence adjacent to the adapter site. A different pool of probes is used to read each base position. The base in that particular location is identified by its fluorescent label. Repeated cycles of ligation build the sequence.
One of the strengths of this sequencing-by-ligation method is that the identification of each base is independent of the identification of its neighbors. “In sequencing by synthesis, if one base is not incorporated, you cannot incorporate the next base. Our approach doesn’t depend on that,” says Radoje Drmanac, chief scientific officer at Complete Genomics, in Mountain View, Calif. “We sequence each base independent of the others. Once we read a base, we reset the DNA back to the original state and start from scratch. Every cycle starts from the same clean conditions.
“This unchained DNA sequencing technique helps improve the quality of the base reads and lower the reagent costs,” Drmanac says. “Each base is equally as good as the first, because they’re independently read,” he says. “We use very low concentrations and small volumes of the labeled reagents, which are the expensive reagents used in every current sequencing technology.”
Applied Biosystems uses chained sequencing by probes that identify two bases at once, and each base is identified by two different probes. This di-base sequencing approach increases the accuracy of the company’s DNA sequences to 99.5%, according to Shaf Yousaf, president of the MCB Genomic Analysis Division at Life Technologies, in Foster City, Calif.
Other sequencing technologies on the horizon require little or no sample preparation, and their developers hope they also achieve longer read lengths.

Companies such as Cambridge, Mass.-based ZS Genetics and San Francisco-based Halcyon Molecular are using electron microscopy as a way to sequence DNA. The companies replicate DNA by using nucleotides modified with heavy-atom labels that can be seen with electron microscopy. “The labels have some real constraints,” says William R. Glover III, CEO of ZS Genetics. “They have to be heavy enough to be detectable, and they have to be light enough not to cause problems with the enzyme. Within that range, they have to be different enough that they can be distinct.”
In these techniques, researchers label and then stretch double-stranded DNA on a surface and take a picture of it with an electron microscope. The position of labels along the DNA molecule reveals the base sequence. “We have molecules that are 10,000 and 20,000 base-pairs long,” Glover says. “We think our read lengths will allow for a coverage ratio of 5 to 8×, which we think will be a huge advantage in reassembly and informatics costs.”
Another technology with potentially long read lengths uses nanopores made from the protein α-hemolysin as the sequencing device. Developed in the lab of Hagan Bayley at the University of Oxford, the nanopores are being commercially developed by Oxford Nanopore Technologies and will be marketed by Illumina.
DNA sequence can be read by the nanopore in one of two ways. In both methods, the bases are discriminated by the change they induce in the amplitude of the current carried by aqueous ions passing through the pore. In one method, an intact DNA strand is threaded through the pore and the bases are identified as they pass a reading head. The challenge with this method is slowing the DNA enough to differentiate the bases as the molecule passes through the pore.
“Over the past year, we’ve made tremendous progress in identifying bases in intact strands of DNA,” Bayley says. Until the problem with DNA speed is solved, Bayley won’t know how long a read length is possible. “There doesn’t seem to be any real reason why you couldn’t feed through tens or even hundreds of kilobases,” he says.
Advertisement
Oxford Nanopore is focusing on the second method, in which an enzyme attached to the pore cleaves bases from the DNA strand so they traverse the pore one at a time. The bases bind to a cyclodextrin molecule attached to the inside of the pore. “The enzyme is going to work at a reasonably slow rate,” thus controlling the problem of excessive DNA speed, says Bayley, who founded the company and serves on its board.
The Philadelphia-based company BioNanomatrix is also working with long DNA strands rather than short fragments. “We are reducing the complexity of the genome analysis problem from one of a 10,000-piece puzzle in assembly and analysis to something more akin to a toddler puzzle of a few pieces,” says Michael Boyce-Jacino, company CEO.
BioNanomatrix’ technology provides access to a source of genetic diversity that is not easily accessed with short-read technologies. These are structural changes in which whole chunks of DNA sequence appear in different places on chromosomes. People are “99.9% identical in DNA sequence, but we’re as little as 85% identical in the organization of our genomes,” Boyce-Jacino says. “We have the same base layout, but the organization is indeed personal.”
Boyce-Jacino compares BioNanomatrix’ technology with Google Maps, in which a user can zoom to different levels of detail. “Currently, DNA sequencing is like going house to house on the map. The problem is that you don’t know if you’re on Elm Street in Ohio or Elm Street in New Jersey. In the genome, because ‘Elm Street’ might be duplicated, you need to be able to zoom out a little bit and understand where that sequence goes to make sure you know where you are in the genome.”
The company’s technology uses a nanofabricated device to separate double-stranded DNA containing 100,000 to 200,000 base pairs into individual lanes. Unique blocks of DNA that are seven bases long are labeled. The location of those blocks within the genome creates a bar code for individual genomes. BioNanomatrix is collaborating with Complete Genomics to use the latter company’s probe ligation approach to sequence short stretches of DNA within the longer stretches. The goal of that long-term project is to reduce the reagent cost of human DNA sequencing to $100 or less per genome.
All of these improvements are accelerating DNA sequencing and lowering its cost. The $1,000 genome is within sight, and the time has come to think about what happens when that barrier has been smashed.
“When we started talking about the $1,000 genome, I don’t think we realized how fast we were going to get there,” says Richard P. Lifton, a genetics professor at Yale University. “We now need to think about when we’re going to get to the $10 or $100 genome. We clearly are headed on a trajectory to be able to do that. We need to think about how we want to deploy the technology.”
You might also like...



The power is now in your (nitrile gloved) hands
Sign up for a free account to get more articles. Or choose the ACS option that’s right for you.
Already have an ACS ID? Log in
Join the conversation
Contact the reporter
Submit a Letter to the Editor for publication
Engage with us on Twitter