Advertisement
Grab your lab coat. Let's get started
Welcome!
Welcome!
Create an account below to get 6 C&EN articles per month, receive newsletters and more - all free.
It seems this is your first time logging in online. Please enter the following information to continue.
As an ACS member you automatically get access to this site. All we need is few more details to create your reading experience.
Not you? Sign in with a different account.
Not you? Sign in with a different account.
ERROR 1
ERROR 1
ERROR 2
ERROR 2
ERROR 2
ERROR 2
ERROR 2
Password and Confirm password must match.
If you have an ACS member number, please enter it here so we can link this account to your membership. (optional)
ERROR 2
ACS values your privacy. By submitting your information, you are gaining access to C&EN and subscribing to our weekly newsletter. We use the information you provide to make your reading experience better, and we will never sell your data to third party members.
Biological Chemistry
First agents to fight Huntington’s enter clinical trials
Previous treatments addressed only disease symptoms
by Erika Gebel Berg, special to C&EN
April 10, 2017
| A version of this story appeared in
Volume 95, Issue 15
If you were at risk for a fatal illness that had no treatment to slow its progress, would you take a test to know your fate? That is a question that people who have a parent with Huntington’s disease must face. Because the neurodegenerative disease is genetic, these people have a 50:50 chance of developing it.
The disease kills brain cells, hindering people’s ability to think and move before eventually taking their lives. Genetic tests for Huntington’s look for a characteristic mutation in the huntingtin gene. In 1993, scientists determined that the mutant version of the gene has extra copies—more than about 38 of them—of the DNA-base-pair repeat sequence CAG. Exactly how Huntington’s arises from this mutation isn’t well understood, but the resulting mutant huntingtin protein forms telltale aggregates in brain cells.
The Huntington’s pipeline
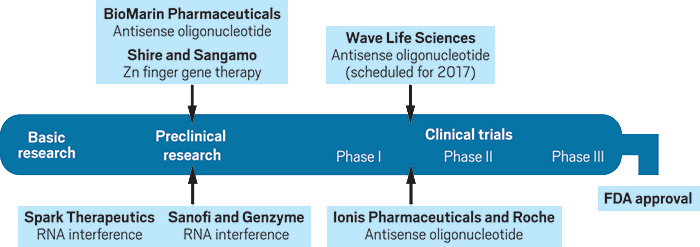
Several companies have disease-modifying treatments for Huntington’s at various stages of the development pipeline.
Credit: C&EN
Only about 5–10% of people at risk for the disease end up getting tested, whereas the remainder wait for symptoms, such as memory lapses and hand trembles, to develop, typically between the ages of 30 and 50. Most people prefer not to know because there isn’t anything they can do about the disease. Current Huntington’s medications don’t slow or alter the progression of the disease; instead, they address only its symptoms.
But that may soon change. By year’s end, results from the first human trial of a disease-modifying Huntington’s treatment are expected, with additional agents entering clinical trials midyear. The data from these trials could provide hope for the more than 200,000 Americans at risk for Huntington’s disease.
Taking down a monster
“When we identified the huntingtin gene, we’d hoped the protein would be something we could inhibit” with a small molecule, says George Yohrling, senior director of mission and scientific affairs at the Huntington’s Disease Society of America (HDSA). “It didn’t turn out to be that way. Huntingtin is a huge, monstrous protein—a scaffolding protein that’s essential for life.” Small-molecule inhibitors are unlikely to block such a big species, so the researchers who developed the treatments that are entering clinical trials turned to silencing the huntingtin gene instead.
These synthetic agents are so-called antisense oligonucleotides: short polymers made up of chemically modified nucleotides. Researchers design the sequence of the nucleotides to complement a portion of a target messenger RNA, in this case the mRNA transcribed from the huntingtin gene.
When these antisense oligonucleotides bind the mRNA, they recruit cellular machinery that tears up the transcript, preventing production of the huntingtin protein. So by targeting the mRNA for the disease-related gene, drug designers can reduce levels of the dysfunctional biological molecule. “You can design an antisense oligonucleotide against any gene,” Yohrling says. “Every gene is druggable now.”
Some experts in the drug development field, however, have questioned the feasibility of antisense oligonucleotide therapies for diseases of the central nervous system, such as Huntington’s. “Antisense oligonucleotides are highly negatively charged,” says David Corey of the University of Texas Southwestern Medical Center. “It was hard to imagine that they could cross the blood-brain barrier and get into the brain, much less find their target. And do all of that without being toxic.” Corey is referring to the cellular barrier that controls which molecules enter the brain and spinal cord from the bloodstream. Highly charged species don’t make the list.
But there are ways around the blood-brain barrier. For example, Ionis Pharmaceuticals developed a drug to treat spinal muscular dystrophy that doctors can deliver via a lumbar puncture directly into the cerebrospinal fluid. The agent, Spinraza, is an antisense oligonucleotide that targets an mRNA in the brain. It was approved by the U.S. Food & Drug Administration in December 2016. The success of Spinraza was a big deal for demonstrating that delivery of antisense oligonucleotides to the brain was possible, Corey says. In fact, the agents furthest along in development for Huntington’s use the same cerebrospinal delivery approach.
Mutant targeting
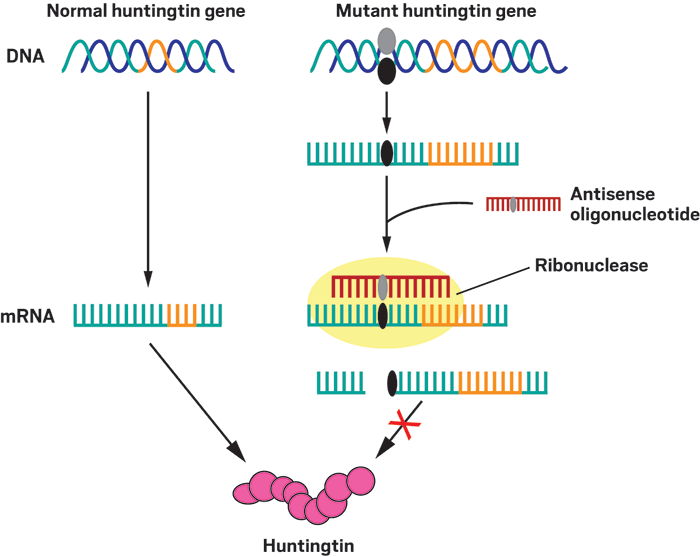
In people with Huntington’s disease, one of their two copies of the huntingtin gene contains extra copies—more than about 38 of them—of the DNA-base- pair repeat sequence CAG (orange). Allele-specific antisense oligonucle- otides (red) selectively bind to the mRNA transcript of this mutant gene by interacting with a sequence that contains a specific single-nucleotide poly- morphism (black and gray ovals) found in the mutant but not in the normal huntingtin gene. When the antisense oligonucleotide binds to the mutant transcript, a ribonuclease enzyme detects the double-stranded nucleotides and cleaves the mRNA strand, preventing its translation into the dysfunc- tional huntingtin protein.
Competing platforms
Ionis is also behind the ongoing Phase I/IIa trial of a Huntington’s antisense oligonucleotide called IONIS-HTTRxthat began in late 2015. Nipping at Ionis’s heels is Wave Life Sciences, a firm that plans to initiate a Phase I/IIa clinical trial of its own antisense oligonucleotide in mid-2017.
The companies’ approaches share some features. Both, for example, depend on ribonuclease H in cells. This enzyme hunts down complexes involving strands of RNA and DNA and then cleaves the RNA strand. Ionis’s and Wave’s antisense oligonucleotides include both DNA and RNA segments to coax ribonuclease H into action.
However, Ionis’s and Wave’s approaches do diverge. One major difference is in the stereochemistry of their oligonucleotides. The chiral center at each linkage between nucleotides can be either right-handed or left-handed. Ionis leaves the stereochemistry at these centers to chance, ending up with a mixture of stereoisomers. But Wave uses a unique synthetic chemistry platform that allows the company to generate stereopure oligonucleotides. “Once we get a sequence we really like, we can do rational drug design” to determine the most effective stereochemistry at each linkage, says Paul Bolno, president and chief executive officer of Wave. A left-handed center could make the oligonucleotide more stable, but a right-handed one may make the agent more potent or less likely to be a target of the immune system, he says.
The two companies also differ in the mRNA transcripts they target. With more than 350,000 nucleotides, the huntingtin transcript is one of the largest in the body, says C. Frank Bennett, senior vice president of research at Ionis. Meanwhile, the antisense oligonucleotides designed to take down this goliath are about 20 nucleotides. Bennett says Ionis researchers screened thousands of oligonucleotides looking for the antisense sequence that best lowered levels of huntingtin in cells and animals. Their approach was agnostic with regard to whether the antisense molecule targeted normal or mutant huntingtin mRNA.
Wave, on the other hand, wanted its oligonucleotide to selectively bind to the mutant transcript. To do so, Wave researchers identified single base-pair differences between normal huntingtin genes and mutant ones outside the regions containing the characteristic CAG repeats. These single-nucleotide polymorphisms (SNPs) act as “evolutionary hitchhikers” on the mutant gene, Wave’s Bolno says. They don’t cause the disease but are associated with the mutations that do.
Wave scientists pinpointed two SNPs that, together, are found in about two-thirds of people with Huntington’s disease. The company designed two antisense oligonucleotides called WVE-120101 and WVE-120102, one for each SNP, and plans to simultaneously run two separate clinical trials, pairing people with a given SNP to the appropriate agent.
Of course, people without one of the two SNPs wouldn’t benefit from the Wave treatments, while the Ionis approach, if successful, should work for anyone with Huntington’s disease. But, HDSA’s Yohrling says, “there might be a role for both approaches,” because even if lowering normal huntingtin is associated with side effects, the treatment may still prolong the lives of people who aren’t candidates for the mutant-specific approach.
Wave went for the allele-specific approach because the firm’s scientists worried that lowering levels of wild-type huntingtin could cause harm. “I think most people would agree that allele-specific is the best and safest way to go,” Yohrling contends. Scientists know embryos require huntingtin for proper development, but its role in adults is less well-defined. Although some studies indicate that huntingtin is important for neuron function, other research suggests lowering levels by about 50% has little effect.
“Ionis considered an allele-specific approach,” Bennett says. “We were confident that targeting in a nonspecific manner would be safe. We’ve looked very thoroughly in our own data and seen no evidence of negative effects.” In Ionis’s tests of mice with Huntington’s-like conditions, the firm’s antisense oligonucleotide lowered levels of huntingtin mRNA by 50–75%, a reduction that was sustained for 12 weeks after the treatment (Neuron 2012, DOI: 10.1016/j.neuron.2012.05.009). Huntingtin protein levels dropped by a similar amount. The treated mice exhibited both delayed onset of disease symptoms and a reversal of motor dysfunction. Additional tests in nonhuman primates, pigs, and dogs by Ionis showed no evidence that reducing wild-type huntingtin, along with the mutant, was harmful.
The ongoing Ionis trial, with support from Roche, involves about 44 adults who have just started exhibiting symptoms of Huntington’s disease. Each patient will receive four increasing single doses of the company’s antisense agent over the course of the trial. With each of the four doses, the researchers will collect spinal fluid to measure the level of mutant huntingtin protein as a biomarker of the candidate therapy’s effectiveness.
Because the duration of the study is short and Huntington’s disease progression can be slow, the researchers don’t expect to observe any clinical benefits. To prepare for longer Phase III trials, though, Ionis’s Bennett says they are also testing cognitive and motor function as clinical markers of disease progression. So far, Ionis reports that no adverse effects have been observed. “Any setback for this study would be a setback for the whole field,” Yohrling says. “While we are very excited about the prospect of huntingtin-lowering drugs in the clinic, there is still a lot to learn.”
Beyond antisense
Although antisense oligonucleotides are the first disease-modifying therapies to reach clinical trials for Huntington’s, the agents are just one of several types in development for the disease. Two companies are working on RNA interference approaches that involve injecting a virus into the brain that would perpetually produce short hairpin RNAs that target the mutant huntingtin mRNA transcript for destruction. Another virus-based strategy would lead to the expression of zinc finger proteins in the brain that act directly on DNA to stop the transcription of the mutant huntingtin gene.
Yohrling says another approach would be to target the aggregated mutant huntingtin proteins. “Could you develop small molecules that induce autophagy—induce the body to selectively degrade mutant huntingtin and aggregated proteins?” he asks. “People are thinking about that, but I think that’s a ways off.”
Despite knowing the precise genetic cause of Huntington’s disease for a quarter century, scientists have struggled to acquire the tools to target the disease mechanism with antisense oligonucleotides. For example, to get to this point, researchers in the field needed to first discover the ribonuclease H degradation pathway and advance synthetic oligonucleotide chemistry. Still, even though the safety and efficacy of these antisense therapies remain unknown, the clinical trials are a true milestone and offer unprecedented hope to people with Huntington’s disease.
Erika Gebel Berg is a freelance science writer based in Washington, D.C.
You might also like...



The power is now in your (nitrile gloved) hands
Sign up for a free account to get more articles. Or choose the ACS option that’s right for you.
Already have an ACS ID? Log in
Join the conversation
Contact the reporter
Submit a Letter to the Editor for publication
Engage with us on Twitter