Advertisement
Grab your lab coat. Let's get started
Welcome!
Welcome!
Create an account below to get 6 C&EN articles per month, receive newsletters and more - all free.
It seems this is your first time logging in online. Please enter the following information to continue.
As an ACS member you automatically get access to this site. All we need is few more details to create your reading experience.
Not you? Sign in with a different account.
Not you? Sign in with a different account.
ERROR 1
ERROR 1
ERROR 2
ERROR 2
ERROR 2
ERROR 2
ERROR 2
Password and Confirm password must match.
If you have an ACS member number, please enter it here so we can link this account to your membership. (optional)
ERROR 2
ACS values your privacy. By submitting your information, you are gaining access to C&EN and subscribing to our weekly newsletter. We use the information you provide to make your reading experience better, and we will never sell your data to third party members.
Materials
The GPU Revolution
Designed for video games, Graphics Processing Units bring once-impossible simulations within reach for chemists
by Lauren K. Wolf
November 1, 2010
| A version of this story appeared in
Volume 88, Issue 44

Three years can pass in a flash for most chemical research projects—and often without yielding three years' worth of useful data. Experiments take time: Techniques need to be carefully developed, conditions tweaked, problems overcome, and promising results verified. Patient chemists involved in the 10- to 15-year drug discovery process can attest to this.
But for computational chemistry, the past three years have released a torrent of data. A revolution in how molecular simulations are carried out is now making it possible for theorists to do speedy desktop calculations that rival those of supercomputers. And it's all thanks to the video-game market.
Consumer demand for lifelike avatars and interactive scenery at a reasonable price have pushed computer firms to come up with relatively inexpensive, yet sophisticated, graphics hardware. Called a graphics processing unit (GPU), this hardware is responsible for three-dimensional images in games. Unlike the central processing unit (CPU) of a computer, which might have a couple of electronic components that carry out mathematics (known as cores), a GPU has hundreds of arithmetic elements that can perform massively parallel calculations.
Computational chemists took note of these graphics cards nearly a decade ago because of their ability to carry out billions of math operations per second, but harnessing their power was tedious and difficult. "In those early days, you really had to represent your calculation as if it were some graphics operation," says Vijay S. Pande, a chemistry professor at Stanford University. In other words, theorists had to jump through a lot of hoops to get the GPUs, which were set up to output shaded polygons, to recognize their algorithms.
It wasn't until 2007, when graphics hardware firm Nvidia introduced Compute Unified Device Architecture (CUDA), a new GPU chip structure and programming tool kit, that things changed. CUDA enabled scientists to access GPUs with high-level programming languages such as C and Fortran, so "it feels like you're writing a more normal computer program," Pande says. Since then, other firms, such as AMD, have followed suit with more-user-friendly GPUs.
These easier-to-use GPUs have transformed the computational field in the past three years. Supercomputers are still at the forefront of computational research (C&EN, Oct. 18, page 5), but when GPUs are incorporated into the clustered machines, more complex calculations become possible. Chemists are now using graphics cards to carry out classical molecular dynamics simulations on desktops, and clusters are beginning to output results on large biomolecular systems that couldn't be easily explored previously. And theorists who do quantum chemical calculations are joining the GPU bandwagon, adapting their more complex algorithms to run on the graphics hardware.
According to Pande, the computational chemistry field is now in the third of a series of evolutionary steps for general-purpose GPU computing. In the first step, just getting a program to run on the GPU at all "was impressive, and usually there was no speedup," he says. In the second, theorists demonstrated some impressive speed increases with GPUs versus traditional CPU-based calculations. But now, Pande explains, scientists are ready to do calculations "that we never even dreamed of doing on CPUs because it would be too slow."
For computational chemists, who are perennially limited by computing power, speed is king. "My mom gives me a hard time and says that I'm just being impatient," says Pande, who has been researching how proteins fold for nearly 20 years. But it currently takes an entire day for a protein-folding calculation to output one nanosecond of simulated folding data for a simple model system. Proteins that are of interest to experimentalists, however, often take a millisecond or longer to fold. "That's about a million times longer," Pande points out, "so it would take a million days, or 3,000 years, which is just ridiculous." GPUs excite the Stanford researcher because they have the potential to reduce calculation time for certain problems from years to months.
Pande is now using the graphics cards in his Folding@home system, a distributed-computing project that links personal computers around the world to a main server and borrows their power to do molecular dynamics calculations. Folding@home, which celebrated its 10th anniversary last month, currently comprises roughly 300,000 to 400,000 machines, about 40,000 of which have GPUs accessible for scientific simulations.
Harnessing these GPUs in the Folding@home system, Pande's group at Stanford recently simulated the 1.5-millisecond folding of a 39-amino-acid fragment of the ribosomal protein NTL9 (J. Am. Chem. Soc. 2010, 132, 1526). The researchers used the molecular dynamics program GROMACS to run thousands of short trajectories that they then pieced together with a network model to achieve the final folding picture. The GPUs were "key" in achieving that final folding trajectory, Pande says.
Even computational scientists who don't require an entire network of computers for their calculations are embracing GPUs. Alex Travesset, a professor in the department of physics and astronomy at Iowa State University, makes do with only one GPU per computer in his group. "You can have a very powerful system by just putting a GPU into an existing computer," he says.
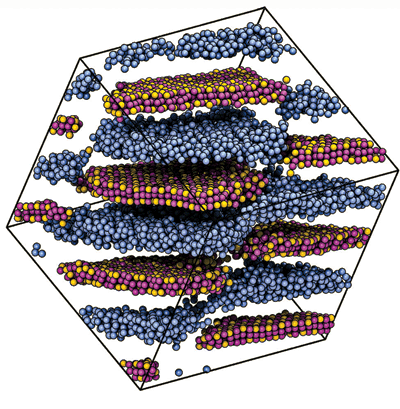
Travesset and his team recently used the GPUs in their lab to systematically simulate the properties of polymer nanocomposites (Phys. Rev. E 2010, 82, 021803). They used molecular dynamics simulations—specifically an open-source molecular dynamics program called HOOMD that was codeveloped by Travesset and former grad student Joshua Anderson—to examine the formation of triblock copolymer-inorganic nanoparticle composites under different conditions of polymer packing, particle attraction, and particle-polymer affinity. In this way, the researchers sampled a vast parameter space and generated phase diagrams for the materials, which might someday be used in solar cells and self-healing coatings.
"We can give a very detailed description of what happens and point out to the experimentalists the region in parameter space where they are going to find some interesting new materials," Travesset says. "We wouldn't be able to get the detailed phase diagrams" if GPUs weren't available.
How scientists determine the exact speedup factor for GPU versus CPU calculations varies. Speedup values, which are typically reported in the 10 to 100 range, depend on the computer systems being compared as well as on parameters such as the size and complexity of the simulation.
Travesset looks at how long a certain molecular dynamics calculation runs on the computer cluster he would use at Iowa State and then looks at the time the same calculation takes on his desktop GPU. "What I see is the following: The 90 cores of this cluster, which is the fastest that we have here, give me the same performance as one Fermi card" he says, referring to the architecture of Nvidia's newest generation graphics card. That's a performance speedup factor of about 90. With a GPU, "I can get it on my own computer right in my office," without having to reserve time on the large public computing system, he adds.
Klaus Schulten, however, prefers making GPU-CPU comparisons directly when possible. "Many people state that they can compute faster on GPUs," but they aren't making proper comparisons, says the director of the Theoretical & Computational Biophysics Group at the University of Illinois, Urbana-Champaign.
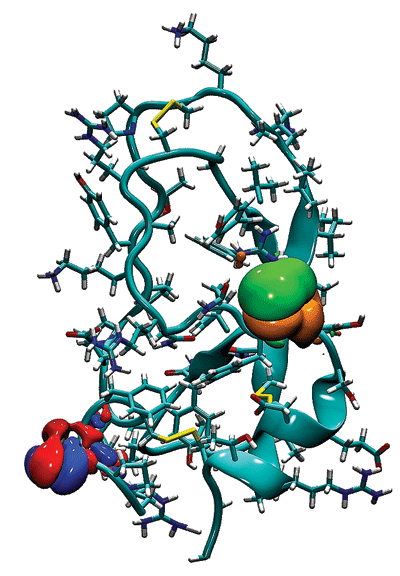
To get what Schulten considers to be an accurate speedup factor, he and his group recently ran some molecular dynamics calculations on a cluster named Lincoln at Illinois' National Center for Supercomputing Applications. They used their own molecular dynamics program, NAMD, to simulate a 3 million-atom system on Lincoln with and without GPUs added to the cluster. "We are opening the cabinet, adding GPUs, closing the cabinet, and running our program," Schulten says.
For this particularly complicated simulation—the ribosome's production of a polypeptide and its subsequent threading through the translocation channel of a cell membrane—the researchers achieved about a fourfold speedup with two GPUs and eight CPUs in the cluster. "This is a realistic speedup" for this type of calculation, Schulten says. To put it in context, an advanced simulation on this system that would normally take two months on a cluster can take just two weeks with the addition of GPUs.
Schulten presented some of these brand-new results in late September at the GPU Technology Conference in San Jose, Calif. The simulations were based on electron cryomicroscopy data for the molecular complex that were obtained by Roland Beckmann, a professor at the Gene Center of the University of Munich.
Where the speedup that GPUs offer becomes vital is in the trial-and-error process that is part and parcel of science. "Maybe you make a mistake, you have to repeat something, or you would like to try out several things," Schulten says. By making simulations faster, "you can be more playful," he says. "Before, you could hardly accomplish one long simulation. Now you can do several."
One reason for Schulten and other researchers' early success in exploring biomolecular systems is that "molecular dynamics is a poster child for an excellent GPU application," says John E. Stone, a senior research programmer who works with Schulten at Illinois. The problem of simultaneously calculating forces on a large number of molecular units—"n-body calculations," in theorist-speak—is ideal for GPUs. The cards are "very good for things that have a regular pattern of computation and that are arithmetic intensive," he says.
In contrast to classical molecular dynamics research, the field of quantum chemistry is not quite far enough along in its incorporation of GPUs to publish application-based papers. "The quantum chemists have it hard" when it comes to GPUs, Stone says. "Their algorithms are very involved, and there are a lot of different ways of doing things." So it's not surprising that many researchers in the community were originally quite reticent to invest time and energy in rewriting their algorithms to run on GPUs.
Mark Gordon, a chemistry professor at Iowa State, did embrace GPUs soon after Nvidia released CUDA and recalls the lack of enthusiasm some of his quantum chemistry colleagues displayed early on. One of the things they worried about was that the first general-computing GPUs couldn't carry out double-precision calculations, he says. Compared with single-precision data, double-precision numbers have more digits to the right of the decimal point, enabling higher accuracy calculations.
"A typical video game has no need for double precision at all," Stone says, so of course the original GPUs didn't include it. Simple molecular dynamics simulations also don't require it. But certain portions of quantum chemical calculations do.
Nvidia and AMD have since released GPUs that have double-precision capabilities, but they are still slower than their single-precision counterparts. "The situation now with GPUs is that if you run in double precision, you take a hit," Gordon says. "You get speedups, but you don't get as good speedups."
As a result, both he and Todd J. Martinez, a chemistry professor at Stanford University whom Gordon calls the GPU "leader" of quantum chemists in the U.S., have investigated which parts of a quantum calculation can be done at lower precision without taking a hit in accuracy. And their groups and others, including that of chemistry professor Alán Aspuru-Guzik of Harvard University, have written GPU-based programs that make use of both precision types by, for example, beginning a calculation in single precision and finishing it in double precision. Martinez' quantum chemistry code, TeraChem, which is customized for doing density functional theory, already runs in mixed precision on GPUs, and Gordon's group is close to getting its quantum chemistry package, GAMESS, running this way as well.
Advertisement
Because redesigning code is a major undertaking, Gordon says his focus thus far "hasn't really been on solving specific chemistry problems as much as it has been on algorithm development." He aims to rewrite the different quantum chemistry functionality codes in GAMESS for GPUs and make them available to the public before moving on to application-based work.
Martinez, however, has begun simulating some molecular systems. "With TeraChem and a GPU, we can now do calculations on up to 50 atoms in real time on a laptop," he says. On a single desktop workstation with eight GPUs running in parallel, he can do even better. Martinez' group, which includes grad student project leader Ivan Ufimtsev, used this setup recently to study protein-water charge transfer in bovine pancreatic trypsin inhibitor—a 2,634-atom system.
The calculation speedups enabled by GPUs can be considered in two ways, Martinez says. "You can either look at this as saying that one can replace clusters with desktops," he says. Or the speedups can be viewed as allowing GPU-based clusters to do more complex calculations, he adds.
Programming GPU clusters, Illinois' Stone says, is one of the next problems to be addressed by computational chemists and computer hardware firms. The way GPUs currently communicate with one another in a cluster is through CPUs, which is time-consuming and inefficient. "You have to work on how to communicate back and forth," GPU to GPU, Gordon contends, if you want to take advantage of extremely parallel calculations on clusters.
According to Stone, GPUs use more power and can generate more heat than do CPUs. But "if you're getting a significant speedup, they're actually more energy efficient for computation than CPUs are," he adds. Scientists interested in building exascale supercomputers—computers capable of completing 1,000 quadrillion floating-point operations per second (flops)—are sure to pay attention to GPU cluster development for this reason.
"A major concern is the power consumption that these huge machines will have," Stone says. They might require hundreds of millions of watts to run—power that would likely need to be supplied by a nuclear reactor. "So you can imagine these scientists," he adds, "are keeping their eyes focused very clearly on what's happening with GPUs."
You might also like...



The power is now in your (nitrile gloved) hands
Sign up for a free account to get more articles. Or choose the ACS option that’s right for you.
Already have an ACS ID? Log in
Join the conversation
Contact the reporter
Submit a Letter to the Editor for publication
Engage with us on Twitter