Advertisement
Grab your lab coat. Let's get started
Welcome!
Welcome!
Create an account below to get 6 C&EN articles per month, receive newsletters and more - all free.
It seems this is your first time logging in online. Please enter the following information to continue.
As an ACS member you automatically get access to this site. All we need is few more details to create your reading experience.
Not you? Sign in with a different account.
Not you? Sign in with a different account.
ERROR 1
ERROR 1
ERROR 2
ERROR 2
ERROR 2
ERROR 2
ERROR 2
Password and Confirm password must match.
If you have an ACS member number, please enter it here so we can link this account to your membership. (optional)
ERROR 2
ACS values your privacy. By submitting your information, you are gaining access to C&EN and subscribing to our weekly newsletter. We use the information you provide to make your reading experience better, and we will never sell your data to third party members.
Business
How phenylketonuria, a once-neglected disease, became a proving ground for new drugs
After decades of yearning for drugs, people with PKU now have 2, and a dozen more are in development
by Ryan Cross
July 8, 2019
| A version of this story appeared in
Volume 97, Issue 27

Credit: Erik Jacobs | The Parrazzo and Ward families, who formed a friendship because they each have kids with PKU, cook a lowphenylalanine dinner at the Parrazzos’ house in Raynham, Massachusetts. Vinny Parrazzo (right) drinks his medical formula before dinner.
It’s dinnertime on a Wednesday evening in June at the Wards’ house in Whitman, Massachusetts. As Karen Ward calls her three kids to the table, her husband, Patrick, prepares a special plate for their 7-year-old. “Lauren, would you rather have more mashed potatoes or more corn?” he asks.
In brief
As far as rare diseases go, phenylketonuria (PKU) is pretty straightforward. It’s caused by mutations in an enzyme that breaks down an amino acid called phenylalanine. But drugs for PKU have been surprisingly hard to make. And for decades, most doctors didn’t think people with PKU needed them—they could prevent the brain damage caused by high phenylalanine levels by simply eating a low-protein diet. But most people have a hard time sticking to the restrictive diet and would like to be able to eat without fear. Today, two proven, but imperfect, drugs are approved for PKU in the US, and a dozen more are in development, covering nearly every drug modality imaginable.
“More corn,” she tells her dad, who begins punching numbers into a calculator. “Fifty divided by 1.75. That’s 28 grams,” he murmurs, while weighing the carefully apportioned amount of kernels on an electronic scale.
While her siblings pack their plates, Lauren gets about a quarter cup of each vegetable. And while the rest of the Ward family eats chicken, Lauren gets a homemade veggie patty, whose ingredients—like the corn—were carefully measured out to the gram. The recipe was supposed to yield six patties, and Patrick pauses as he realizes he made only five. “We’ll have to divide,” he says, grabbing the calculator and craning back to study the recipe. “It’s like an algebra class every night.”
These mathematical acrobatics are the norm for the Wards, and for the many families who have a child with the rare disease called phenylketonuria, or PKU. Lauren was born with mutations in a critical metabolic gene that carries the recipe for making an enzyme called phenylalanine hydroxylase (PAH). Those mutations prevent PAH from doing its job of breaking down phenylalanine, one of the 20 amino acid building blocks of proteins.
If Lauren eats too much protein, high levels of phenylalanine will accumulate in her brain, making it hard for her to think clearly and, over time, causing severe intellectual disability. Dairy, eggs, and meat are obviously off limits. Most flour-based foods, including pasta, have too much protein for someone with PKU. Even dietary staples like corn and potatoes have enough phenylalanine that they can be consumed only in limited quantities. That’s why Patrick and Karen spend each meal carefully measuring Lauren’s portions.
When Lauren was born, there was just one drug for PKU. Now there are two, both sold by BioMarin Pharmaceutical. The first, called Kuvan, loosens the restrictive diet for a fraction of people with the disease, including Lauren, allowing them to eat a few extra grams of protein each day. The second, Palynziq, is a daily injection of phenylalanine-degrading enzymes that allow people with PKU to grab whatever they want out of the fridge. But Palynziq is approved only for adults, and the prescription comes with a warning about potentially severe immune reactions that hospitalized some people during clinical trials for the drug.
Soon, people with PKU could have more and better options. Seeing Palynziq’s impending approval, a dozen biotech companies began taking nearly every conceivable approach to tackle PKU: once-a-day pills that stabilize the mutant enzyme, cells transformed into phenylalanine-munching machines, and a plethora of gene-therapy approaches promising a permanent cure are all on the table.
It’s a curious situation for a disease that affects only about 16,000 or so people in the US. But there are many reasons to pick PKU, explains Arthur Tzianabos, CEO of Homology Medicines, which is developing a gene therapy for PKU. To start, PKU’s cause is well understood, and as rare genetic diseases go, PKU is one of the most common. Furthermore, BioMarin gave competitors a road map to follow and goals to beat. And since measuring phenylalanine levels in the blood is easy, it shouldn’t take long for firms to know if their experimental medicines are working.
Some of the companies pursuing PKU have loftier ambitions. The rare disease offers the potential to quickly assess the safety and effectiveness of their technologies before unleashing them on other, potentially more lucrative diseases. “All those factors led companies to pick PKU if they had a platform they wanted to prove out,” Tzianabos says.
If even a few of them are successful, the protein-counting days of families like the Wards could be numbered.
An enzyme substitute
Before the introduction of the phenylalanine-restricted diet in the 1950s and ’60s, children with PKU became severely intellectually disabled and, often, institutionalized. Then, the 1963 arrival of a heel-prick blood test for high phenylalanine levels meant babies with the disease could be identified soon after birth and put on the special diet immediately.
“The dogma was that PKU treatment had been solved,” says Cary Harding, a metabolic disease specialist at Oregon Health & Science University. “But if you came in and started treating patients, it was obvious that things had not been solved.”

Credit: Erik Jacobs
Karen Ward (right) gives her daughter Lauren (middle) her medical formula before dinner. The drink contains the 19 amino acids besides phenylalanine, and many of her calories for the day.
A survey of 625 people conducted by the National PKU Alliance in 2015 found that more than 60% of adults and 25% of children with PKUcan’t keep their phenylalanine levels in check. Elevated levels cause brain fog, which some people with PKU describe as a hangover without the nausea. Many of them have difficulty following multistep instructions, reading books, and staying employed. Anxiety and attention deficit disorder are common among people with PKU, too.
Lauren seems to be on track in school, but sometimes she can be forgetful, Karen Ward says. “I don’t know if it is just her age, or PKU related.”
Surprisingly, even with all that researchers know about the mechanics of the disease, doctors still aren’t sure why high levels of phenylalanine damage the brain.
The dogma
was that PKU
treatment had
been solved.
Cary Harding, metabolic disease specialist, Oregon Health & Science University
In theory, once the genetic defect was identified in the 1970s, PKU should have been an easy fix. After all, several biotech companies, including BioMarin, have come up with ways to replace missing or broken enzymes for other metabolic diseases. But PAH is particularly complicated. Four copies of the enzyme must coalesce into a butterfly-like structure called a tetramer, and two cofactors—oxygen and a molecule called tetrahydrobiopterin—must be present before PAH will start breaking down phenylalanine. Simply giving people PAH didn’t work for PKU.
So scientists decided to try a PAH substitute: a plant enzyme called phenylalanine ammonia-lyase (PAL), which also breaks down phenylalanine. In 1999, BioMarin acquired a small firm that invented a PAL enzyme substitution therapy designed to be taken orally and break down phenylalanine in the gut. But in mice studies, the enzyme just got digested. BioMarin then tried injections of the drug, but the immune system saw the plant protein as an invader and neutralized its effect.
BioMarin recruited scientists from McGill University and Scripps Research to locate better versions of the PAL enzyme and chemically modify it to subdue the immune reaction it caused. Human studies of the modified enzyme, now derived from bacteria, finally began in 2008, and it took another decade for regulators to approve the drug, Palynziq, in the US.
All told, BioMarin spent $618 million on Palynziq R&D over nearly 2 decades. With an average list price of $267,000 per year, Palynziq could eventually bring the company more than $1 billion in annual sales.
The Wards, along with many other PKU families, are happy to see Palynziq finally approved, but they’re not thrilled about the idea of their daughter taking it. For some people in the trial, the foreign enzyme caused such severe anaphylaxis, a strong immune reaction, that they were briefly hospitalized. Many others temporarily developed full body hives and joint aches. And women have to stop taking the drug and return to their low-protein diets if they want to become pregnant.
“I’m not sure that giving yourself an injection every day and risking those side effects are something I’d be willing to take on,” Karen Ward says. But since Palynziq was approved only for adults, and specifically those with high, unmanaged phenylalanine levels, Lauren won’t be trying it anytime soon.
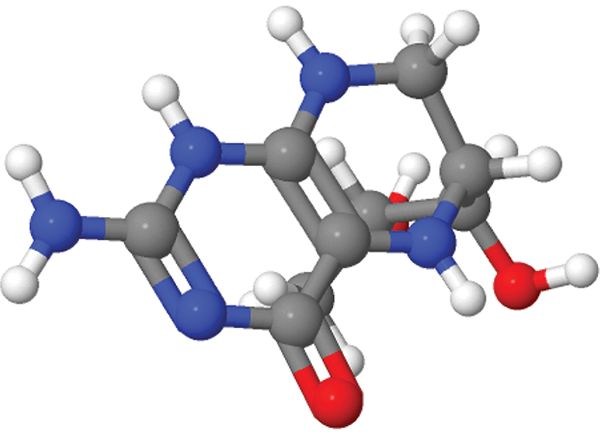
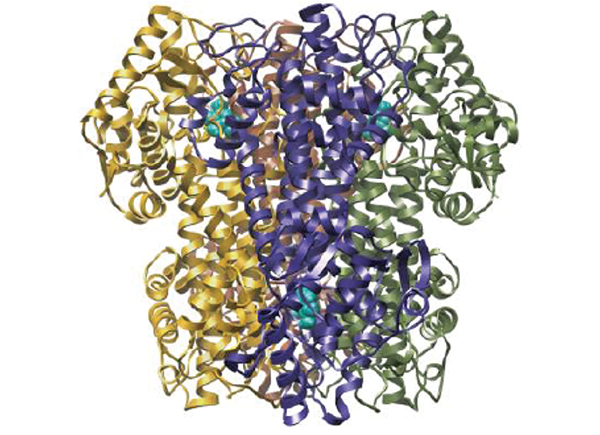

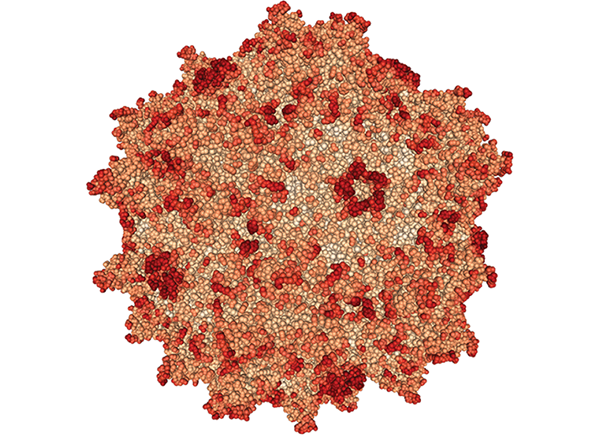
Credit: Biological Magnetic Resonance Data Bank (cofactor), National PKU Alliance (enzyme), RCSB Protein Data Bank (virus), Pixabay (blood), Shutterstock (gene editing, microbes, mRNA, and small molecules
More than one way to treat a disease
PKU is caused by mutations in the gene that makes an enzyme called phenylalanine hydroxylase (PAH). Drug companies have devised numerous ways to tackle the same problem, but all involve either restoring or stabilizing the natural PAH enzyme or supplying a similar enzyme called phenylalanine ammonia-lyase (PAL), which is normally found in bacteria and plants.
PKU is caused by mutations in the gene that makes an enzyme called phenylalanine hydroxylase (PAH). Drug companies have devised numerous ways to tackle the same problem, but all involve either restoring or stabilizing the natural PAH enzyme or supplying a similar enzyme called phenylalanine ammonia-lyase (PAL), which is normally found in bacteria and plants.
Other enzyme approaches
Inside a suburban house just a 30 min drive south of the Wards, another mealtime weigh-in of food will soon be underway. Dinner hasn’t started yet at the Parrazzos, so 20-year-old Anthony Parrazzo; his 14-year-old sister, Samantha; and his 11-year-old brother, Vinny, are sipping on their medical formula, which contains the other 19 amino acids their bodies need besides phenylalanine, plus most of their calories for the day.
Tanya and Vinny Parrazzo have four kids, and three of them were born with PKU. Since they’re allowed to eat only about 3 g of protein per day, the family goes through a lot of salad. “Today I had a family-size salad kit, which is a tremendous achievement,” Anthony jokes.
The Parrazzos’ cupboards and freezers, like the Wards’ home, are filled with expensive, low-phenylalanine foods that no one is thrilled about. A box of special spaghetti sets them back $11.49. A bag of low-protein bagels costs $13.49. And an artificial egg mix rings in at $16.99. Anthony likes the pasta, and Vinny prefers the bagels. Samantha usually sticks to salads.
The state of Massachusetts reimburses up to $5,000 worth of medical foods, and the Wards’ and Parrazzos’ insurance covers their kids’ calorie-packed medical formula, which otherwise can cost $10,000 a year. Yet those bills will surely pale in comparison to the price tags of the high-tech therapies that could one day allow the Parrazzo kids to eat whatever they want.
Many of those therapies are being developed just a short commute away, in Cambridge, Massachusetts. Researchers at Rubius Therapeutics, for instance, are inserting genes encoding the PAL enzyme into lab-grown human red blood cells, turning them into little machines that dismantle phenylalanine in the bloodstream. Like Palynziq, the cell therapies are designed to allow people with PKU to have a normal diet, but with injections on the order of weeks or months instead of every day. “Personally, I’m not interested in marginal improvements,” says Rubius CEO Pablo Cagnoni. “Our goal here is to completely change the lives of PKU patients.”
In March, the company got approval from the US Food and Drug Administration to run a Phase Ib study in 10 adults with PKU. The goal is to evaluate the approach’s safety and determine how long the cells survive in humans. Results from that trial will be reported later this year.
Rubius is one of three companies piggybacking on the PAL approach. Another Cambridge-based biotech, called Synlogic, is installing PAL and another phenylalanine-degrading enzyme into bacteria. The hope is to make probiotic-like therapies, likely taken three times a day during meals; the therapies would break down phenylalanine in the gut. The company has finished one safety study in healthy people, and it plans on sharing data from an ongoing trial in adults with PKU this summer.
A third company, Codexis, used a technique called directed evolution to take another shot at developing PAL enzymes that can be swallowed to break down phenylalanine in the gut. Promising but unpublished results from multiple animal studies and healthy human volunteers led Nestlé Health Science to license the program from Codexis in February.
All three of these biotech businesses are built around unproven technologies, so it makes sense that they would pick a well-understood disease like PKU to test out their approaches. “It was very clear what you needed to do,” says Caroline Kurtz, who leads Synlogic’s metabolic programs. “This disease was the perfect place to start for a platform company like ours.”
It may seem that PKU has become the guinea pig for testing out new ideas in the drug industry. Even if that’s true, most PKU families, including the Parrazzos and the Wards, don’t feel that way. “I kind of wish they’d all get together and just make one thing,” Patrick Ward says. “But I’m just excited that they are actually trying.”
Boosting the enzyme they have
For parents who have kids with PKU, the next meal is always on their minds. The Wards keep a notebook on their kitchen counter to track the milligrams of phenylalanine that Lauren eats—her doctors have determined that she should get 350 mg of the amino acid each day. On that June night, Lauren has had 150 mg, so Patrick is trying to measure out 200 mg more between the corn, potatoes, and veggie patty. “It’s not like golf where you want the lowest number,” he says. “You have to try to hit that number.” If Lauren doesn’t eat enough, her body might start breaking down its own muscle, leading to a harmful spike of phenylalanine in her blood.
Advertisement
Current medical guidelines suggest that people with PKU keep their blood phenylalanine levels below 360 µmol/L. Exactly how much dietary protein that translates to is different for every person. Scientists have documented more than 1,000 mutations in the PAH enzyme so far. On her own, Lauren can safely eat 4 g of protein a day, and with the help of BioMarin’s Kuvan, she can eat 7 g—equivalent to about 3 tablespoons of pinto beans.
Unlike the PAL-based therapies, which in theory should work for everyone, Kuvan works for only about 20–30% of people with PKU. It’s a synthetic version of the tetrahydrobiopterin cofactor required by the PAH enzyme. Boosting its levels seems to stabilize certain forms of the mutant enzymes.
The Parrazzo kids all use Kuvan too—Anthony takes 11 pills a day—although Tanya says it doesn’t help them eat extra protein. They tried the drug on Anthony when he was in third grade because he was daydreaming a lot in class. “When he went on Kuvan, it seemed to help him focus a little bit more,” Tanya says.

Credit: Erik Jacobs
Anthony Parrazzo (center) counts out 11 pills of Kuvan, which he takes every day to help manage his PKU. Lauren Ward (left) and Samantha Parrazzo (right) take a powdered form of the drug.

Credit: Erik Jacobs
Karen Ward weighs a low-protein pasta made from cornstarch and potato flour for her daughter Lauren so that she can calculate how many grams of phenylalanine it contains.
It’s prescribed as a pill or a powder, which makes it easy for Lauren to take. And that’s a top priority for the Wards. “Even though the diet is strict and difficult to manage, Lauren has a good quality of life,” Karen says. “So the biggest thing I would want to see in a new drug is the ease of taking it.”
At least two companies are now working on new small molecules that give PAH a boost but work more potently and for more people than Kuvan.
Karen has visited one of those companies, Cambridge-based Agios Pharmaceuticals, to help educate the firm on what it’s like to raise a kid with PKU. Agios, best known for its drugs that target metabolic pathways in cancer, was “looking for a rare genetic disorder to leverage our metabolism expertise,” Chief Scientific Officer Scott Biller says.
Agios scientists realized that the majority of PAH mutations appear to prevent the enzyme from folding into its proper structure. “Our hypothesis is that when the mutant protein is being made, it never gets to the tetramer,” that butterfly-like shape it must form to properly function, Biller says. The firm has developed a drug candidate that stabilizes some mutant forms of PAH in mouse models. “We don’t expect to treat every single patient,” Biller concedes, but he thinks it will work for the most severe forms of PKU. Agios has not yet disclosed a timeline for when that program will be advanced to clinical trials.

Credit: Erik Jacobs
Patrick Ward uses an app to calculate how many milligrams of phenylalanine are in the zucchini to be served with dinner.
Another Cambridge-based company, called Camp4, has also chosen PKU for one of its lead programs. The start-up specializes in finding targets that can be drugged to increase or decrease the production of particular proteins. Camp4 won’t disclose specifics yet, but CEO Josh Mandel-Brehm says the firm has a small molecule that boosts the expression of two genes and lowers blood phenylalanine levels in mice.
It’s hard to believe that there will be room for multiple small-molecule approaches to compete with the mosh pit of modalities in play to treat PKU. But Mandel-Brehm is confident about his PKU program. “We think it is a relatively big market,” he says.
Biotech firms estimate that the market for PKU is about 50,000 people worldwide. And 91% of people who took the National PKU Alliance’s survey said that development of new therapies was important to them. Christine Brown, the association’s director, knows this firsthand. She has two sons with PKU, and while her 13-year-old is ready to try Palynziq once he’s old enough, his younger brother can’t stand the idea of getting a shot every day. “We are looking forward to the day when people have more options,” Brown says. “We want new treatments, and we want a cure.”
Chasing a cure
Today, no fewer than seven companies, BioMarin included, are working on gene therapies for PKU. Although their approaches differ slightly, they all have the same goal: give people fully functional copies of the PAH gene, potentially providing a onetime, permanent fix. “The bottom line is that to cure this disease, you need to put the natural enzyme back,” Homology’s Tzianabos says.
Getting the DNA that encodes that enzyme into the body is the tricky part. In mouse studies, BioMarin, Homology, Pfizer, Sanofi, and Ultragenyx Pharmaceutical have all used hollowed-out adeno-associated viruses (AAVs) to shuttle the PAH gene to liver cells, where phenylalanine is normally broken down. So far, the animal studies look promising.
“I think gene therapy sounds great,” Karen Ward says, but she is worried about its effect fading over time. That concern is echoed across PKU families. The thought of having a normal, unrestricted diet for 5, 10, or 20 years only to have to go back to their old protein-counting ways horrifies people with PKU.

Credit: Erik Jacobs
After Lauren (center, front) was born with PKU, the Ward family (from left: Katie, Patrick, Karen, and Matthew) had to adjust their lifestyle to meet her dietary restrictions. “It has definitely put a halt to going out for dinner as a family,” Karen says. “In our society, everything is about food. I don’t think you can fully understand how difficult the diet is.”
Another problem is that gene therapy would probably be useful only for adults. Since the liver keeps growing until adulthood, companies are worried that gene-therapy DNA may quickly become washed out in children as their liver cells divide and grow. And once someone has received an AAV-based gene therapy, they develop antibodies against the viruses and can’t get the treatment again. Furthermore, some people are already immune to the viruses and ineligible to get gene therapies in the first place.
This disease was
the perfect place to
start for a platform
company like ours.
Caroline Kurtz, head of translational sciences and product development, Synlogic
Several companies have plans to circumvent AAV’s problems. Much like the PAL-based companies, these biotech firms are using PKU as a proving ground for their technology.
“If there had been a viable cure on the horizon, we wouldn’t have been as excited about this particular disease,” says Jeffrey Galvin, CEO of American Gene Technologies. His company is developing a PKU gene therapy with a lentivirus, which, unlike AAV, integrates itself into the human DNA. That approach could solve the problem of the gene therapy washing out in children, but it has previously caused some people to develop leukemia. Since then, scientists have learned how to more safely use the virus, he says.
The start-up Generation Bio is working on a new way to safely cure children of PKU by using lipid nanoparticles, rather than viruses, to deliver DNA. CEO Geoff McDonough hopes the technique will avoid AAV’s immune problems, allowing the company to give the gene therapy to infants soon after birth and readminister it as needed throughout childhood and adolescence to keep the liver filled with working PAH enzymes.
Some scientists are studying the potential of gene editing for PKU as well. One lab has even used CRISPR base editors to fix single-letter mutations in a mouse model of PKU. But since there are hundreds of ways PAH can be mutated, the base-editing approach would be impractical for treating large numbers of people with PKU. Homology has plans for a second PKU program to use its viruses to precisely slide a full copy of the PAH gene into a person’s DNA, which it considers to be another kind of gene editing.
The limitations of gene therapies may make room for an alternative approach that uses mRNA, the intermediate code between the DNA of genes and proteins, to deliver instructions for making the PAH enzyme to cells. The mRNA-therapy company Moderna has a program for PKU that could enable the body to produce PAH enzymes for a moderate amount of time, maybe weeks or months, before another dose is needed. It’s essentially a temporary version of gene therapy.
Moderna has a giant pipeline of 21 programs, and PKU isn’t a front-runner. The firm is focusing on another, deadlier metabolic disorder called methylmalonic acidemia first. But Paolo Martini, Moderna’s chief scientific officer for rare-disease research, says that if mRNA therapy for that disease works, “then obviously all the other metabolic disorders will be pretty straightforward.”
Yet even for a disease as seemingly simple as PKU, there is no single drug modality that’s clearly better than the others. For now, every one has its pros and cons. The fact that companies are developing so many different technologies to tackle the same problem is a testament to the biotech industry’s growth.
Ten years ago, gene therapy was still viewed as unsafe, Martini says. Engineered cell therapies, gene editing, and mRNA therapy were hardly even on the radar, he adds. “Now we have all of these technologies, and that’s why we can go after PKU.”
Advertisement
Despite the large number of gene-therapy companies working on PKU, the Parrazzos and Wards don’t spend much time talking about a cure. They’re informed enough to know that gene therapy is still an unproven technology for PKU, and they’d be happy with a drug that alleviates the strict diet, like Palynziq, but without the side effects. “PKU is not necessarily an easy thing,” Tanya Parrazzo says. “But it’s what we know.”
Tanya’s kids all want something different from future therapies. Samantha says that if she knew gene therapy worked in humans, she would try it. Her younger brother, Vinny, thinks it’s too risky. Anthony, the oldest, isn’t holding his breath for any new drug. “I really don’t care about being totally PKU-free,” he says
Samantha can’t relate. “Why would you not want it cured?” she asks.
There are certain foods that Anthony wishes he could eat—cheese lasagna comes to mind—but he’s grown accustomed to his lifestyle. “And it’s been working,” he says. “So why change? Why fix something that’s not exactly broken?”
Anthony knows he’s in the minority. Most people with PKU—people like his sister—would love more options.
That so many options are on the horizon “makes me feel like we’ve made PKU heard and known,” Karen Ward says. And that’s a good feeling to have. “It’s a very exciting time for PKU.”
CORRECTION
The story was modified on July 8, 2019 to note that Agios has not yet disclosed when its small-molecule PKU program will advance to clinical trials.
Chemical & Engineering News
ISSN 0009-2347
Copyright © 2024 American Chemical Society
You might also like...



The power is now in your (nitrile gloved) hands
Sign up for a free account to get more articles. Or choose the ACS option that’s right for you.
Already have an ACS ID? Log in
Join the conversation
Contact the reporter
Submit a Letter to the Editor for publication
Engage with us on Twitter