Advertisement
Grab your lab coat. Let's get started
Welcome!
Welcome!
Create an account below to get 6 C&EN articles per month, receive newsletters and more - all free.
It seems this is your first time logging in online. Please enter the following information to continue.
As an ACS member you automatically get access to this site. All we need is few more details to create your reading experience.
Not you? Sign in with a different account.
Not you? Sign in with a different account.
ERROR 1
ERROR 1
ERROR 2
ERROR 2
ERROR 2
ERROR 2
ERROR 2
Password and Confirm password must match.
If you have an ACS member number, please enter it here so we can link this account to your membership. (optional)
ERROR 2
ACS values your privacy. By submitting your information, you are gaining access to C&EN and subscribing to our weekly newsletter. We use the information you provide to make your reading experience better, and we will never sell your data to third party members.
Mass Spectrometry
Fragment correlation mass spectrometry: a new approach to analyzing tandem mass spectra
Using noise in their signal can reveal product ions’ shared precursor
by Fionna Samuels
August 13, 2024
| A version of this story appeared in
Volume 102, Issue 25
Tandem mass spectrometry is a powerful tool in proteomics. By ionizing peptides and then fragmenting those ions into smaller product ions, researchers can determine the biomolecules’ amino acid sequences. But if a sample contains a complex mixture of peptides, it can be challenging to determine which precursor produced which product ions.
To avoid such confusion, scientists have implemented other analytical approaches—such as liquid chromatography—to physically separate peptides prior to injection into the mass spectrometer. But, trying to fully separate similar peptides using physical approaches can prove futile, and the resulting mix of product ions remains difficult to interpret.

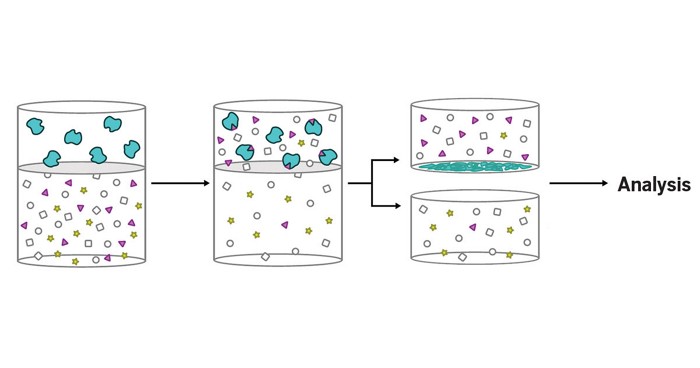
Credit: Yangjie
Li/Stanford
When two different ionized peptides fragment into product ions, some of those ions might have the same mass-to-charge ratio, or m/z. Fragment correlation mass spectrometry differentiates these isobaric ions by grouping them with other ions arising from the same fragmentation pathway (green or blue).
Now, an interdisciplinary team of researchers has demonstrated a way to overcome the challenge: use a new technique—dubbed fragment correlation mass spectrometry—on a mix of parent peptides and group product ions based on the noise in their respective signals (Proc. Natl. Acad. Sci. U.S.A. 2024, DOI: 10.1073/pnas.2409676121).
The approach relies on the innate relationship between product ions fragmented from the same peptide, says lead author Yangjie Li, a postdoctoral scholar and chemist at Stanford. Because these ions arise from the same fragmentation pathway, they will share the same pattern of statistical variation, or noise, in their signal.
When the team performs multiple mass scans, their software can find these patterns and use them to identify ions from the same precursor.
“In proteomics, that’s a big step to fragment everything all at once rather than sequentially fragment one molecule at a time,” says Joseph Loo, a biochemist from the University of California, Los Angeles. Not only does the technique separate ions with the same mass-to-charge ratio, but it can also trace internal fragment ions back to a parent peptide—a capability that caught Loo’s eye.
“Internal fragment ions are a result of multiple cleavage steps in a fragmentation experiment,” he explains, making them incredibly difficult to assign. This new approach might make it much easier.
The currently available code is optimized for the mass spectrometer setup used by Li and her colleagues. If a scientist wants to try it out on their own samples, they will need to reach out to the research team for guidance. But once it’s modified to be easily implemented by those using different setups, Loo says, “all of a sudden, everyone will be doing this.”
When that happens, he won’t be surprised if the method unveils interesting new information in scientists’ proteomic data sets.
Chemical & Engineering News
ISSN 0009-2347
Copyright © 2024 American Chemical Society
You might also like...


Advertisement
The power is now in your (nitrile gloved) hands
Sign up for a free account to get more articles. Or choose the ACS option that’s right for you.
Already have an ACS ID? Log in
Join the conversation
Contact the reporter
Submit a Letter to the Editor for publication
Engage with us on Twitter