Advertisement
Grab your lab coat. Let's get started
Welcome!
Welcome!
Create an account below to get 6 C&EN articles per month, receive newsletters and more - all free.
It seems this is your first time logging in online. Please enter the following information to continue.
As an ACS member you automatically get access to this site. All we need is few more details to create your reading experience.
Not you? Sign in with a different account.
Not you? Sign in with a different account.
ERROR 1
ERROR 1
ERROR 2
ERROR 2
ERROR 2
ERROR 2
ERROR 2
Password and Confirm password must match.
If you have an ACS member number, please enter it here so we can link this account to your membership. (optional)
ERROR 2
ACS values your privacy. By submitting your information, you are gaining access to C&EN and subscribing to our weekly newsletter. We use the information you provide to make your reading experience better, and we will never sell your data to third party members.
Synthesis
Amination Advance
New reaction is first to catalytically convert allylic C-H to C-N
by Stu Borman
June 4, 2007
| A version of this story appeared in
Volume 85, Issue 23
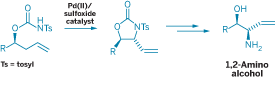
MAKING CERTAIN antibiotics and other nitrogen-containing compounds has just gotten easier with the solution to a long-standing problem in organic synthesis.
Chemists have sought a direct catalytic way to replace hydrogen with nitrogen at an allylic carbon, the sp3-hybridized carbon atom adjacent to a C=C group, a common target of modification in organic compounds. So far, the primary means for putting nitrogen at that carbon requires going through oxygen. Typically, a hydroxyl group replaces the hydrogen first and is then replaced by a nitrogen-containing group. This route can lengthen and complicate syntheses, because the oxygenated materials require additional manipulations.
Now, the oxygen-involving step can be sidestepped. Chemistry professor M. Christina White and grad student Kenneth J. Fraunhoffer at the University of Illinois, Urbana-Champaign, have devised the first method that catalytically converts an allylic C-H directly to C-N (J. Am. Chem. Soc. 2007, 129, 7274).
"This discovery redefines the state of the art for making certain types of molecules," White says. The reaction makes it possible to more rapidly synthesize stereochemically defined oxazolidinones, which form the skeletons of a class of antibiotics that includes linezolid. Oxazolidinones also can be further transformed into medicinally important amino alcohols.
White and Fraunhoffer used the reaction to create a key chiral oxazolidinone intermediate in the synthesis of acosamine, an amino sugar found in the cancer drug epirubicin. They needed only half the number of steps required by the conventional synthetic route, which goes through oxygen.
The reaction uses a Pd(II)/sulfoxide catalyst to transfer nitrogen in an N-tosyl carbamate group directly to an allylic carbon atom.
For now, White's group has demonstrated only an intramolecular version of the reaction, in which the carbamate is tethered to the allyl group. Tethering, White says, increases the reactivity of the process and promotes diastereoselectivity. But tethering isn't a necessary condition for the reaction, and White's group is currently working on a more versatile intermolecular version.
The main challenge in developing the reaction "was to avoid a competing reaction of nitrogen directly with the olefin," White says. The Pd(II)/sulfoxide catalyst system they used avoids this pitfall, because it preferentially activates the allylic C-H bond for cleavage (thereby making this carbon accessible to nitrogen attack), whereas other catalysts of this type tend to make the olefin receptive to nucleophilic attack by nitrogen.

White notes that the new reaction complements a set of rhodium nitrene-based aliphatic C-H aminations developed recently by Justin Du Bois' synthetic chemistry group at Stanford University. Together, the two types of reactions "begin to create a toolbox of C-H-to-C-N bond-forming reactions that have significant potential for streamlining the process of small-molecule synthesis," White says.
Her technique "is significant, as the novel functionalization of allylic C-H bonds allows access to 1,2-amino alcohols, which are key components of nonnatural amino acids and amino sugars," comments synthetic chemist Reinhard W. Hoffmann of Philipps University, in Marburg, Germany. It also makes it possible to introduce "functionality late in a synthesis on elaborate molecular skeletons, and it is likely, while not yet demonstrated, that it does not require protection of bystander functional groups. Hence, there is the clear potential of streamlining overall syntheses of complex target structures."
Synthetic chemist Phil Baran of Scripps Research Institute says the new reaction "is a breakthrough" that "will find widespread use in total synthesis. It compels us to look at the allylic C-H bond in an entirely new way and will have a dramatic impact in the way we think about making molecules."
You might also like...



The power is now in your (nitrile gloved) hands
Sign up for a free account to get more articles. Or choose the ACS option that’s right for you.
Already have an ACS ID? Log in
Join the conversation
Contact the reporter
Submit a Letter to the Editor for publication
Engage with us on Twitter