Advertisement
Grab your lab coat. Let's get started
Welcome!
Welcome!
Create an account below to get 6 C&EN articles per month, receive newsletters and more - all free.
It seems this is your first time logging in online. Please enter the following information to continue.
As an ACS member you automatically get access to this site. All we need is few more details to create your reading experience.
Not you? Sign in with a different account.
Not you? Sign in with a different account.
ERROR 1
ERROR 1
ERROR 2
ERROR 2
ERROR 2
ERROR 2
ERROR 2
Password and Confirm password must match.
If you have an ACS member number, please enter it here so we can link this account to your membership. (optional)
ERROR 2
ACS values your privacy. By submitting your information, you are gaining access to C&EN and subscribing to our weekly newsletter. We use the information you provide to make your reading experience better, and we will never sell your data to third party members.
Physical Chemistry
The Chemical Bond
Whether it's sextuple bonds or bonds involving no shared electrons, chemists chase down new modes of bonding
by Stephen K. Ritter
January 29, 2007
| A version of this story appeared in
Volume 85, Issue 5
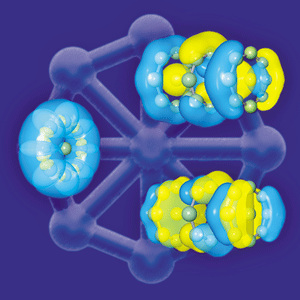
"SOMETIMES IT SEEMS to me that a bond between two atoms has become so real, so tangible, so friendly, that I can almost see it. Then I awake with a little shock, for a chemical bond is not a real thing. It does not exist. No one has ever seen one. No one ever can. It is a figment of our own imagination."
This quote conveys the words of Charles A. Coulson, a prominent theoretical chemist at the University of Oxford, who influenced a generation of chemical-bonding enthusiasts from the 1940s to the early 1970s. Chemistry Nobel Laureate Roald Hoffmann of Cornell University likes to recite Coulson's comments from the early 1950s as part of his own lectures on chemical bonding.
Hoffmann knows something of bonding, having devoted much of his career to being intimate with molecular orbitals. With Robert B. Woodward at Harvard University in the early 1960s, Hoffmann helped develop the famous Woodward-Hoffmann rules that predict the stereochemistry of certain organic reactions.
"There is nothing more fundamental to chemistry than the chemical bond," Hoffmann says. "But it's not a simple concept." He has attempted to explain why during talks on "all the ways to have a bond" at several venues in the past couple of years.
Hoffmann advises chemists to "push the concept of the bond to its limits and accept that, at the limits, a bond will be a bond by some criteria, but not by all." He also advises chemists to have fun with the richness of bonding precisely because it's not clearly defined.
And that is what the international community of computational chemists has been doing. New ways of thinking about standard bonding concepts keep cropping up. Meanwhile, new modes of chemical bonding continue to be reported.
Some examples include bonds in which there are no shared electrons between atoms and, at the other extreme, a molecule with a sextuple bond. Other topics attracting attention include the molecular interactions that give rise to the reactivity of metalloenzymes; aromaticity in metal and nonmetal clusters; and bonding in new materials such as nanoparticles, molecular magnets, and quantum dots.
Bonding is in the midst of a renaissance, according to Gernot Frenking of Philipps University, in Marburg, Germany, and Sason Shaik of Hebrew University of Jerusalem, in Israel. These leading computational chemists recently teamed up to produce a special issue of the Journal of Computational Chemistry (JCC), published on Jan. 15, dedicated to chemical bonding. The issue is also dedicated to the late Gilbert N. Lewis of the University of California, Berkeley, who is considered the father of the chemical bonding concept. The nearly 500-page special issue includes dozens of essays and articles on the history of chemical bonding research as well as the area's leading edges; it is available online at www3.interscience.wiley.com/cgi-bin/jissue/113493174.
In one of the essays, Shaik notes that the "electronic structure revolution" leading to the current descriptions of chemical bonding started 90 years ago when Lewis published a seminal paper entitled "The Atom and the Molecule" (J. Am. Chem. Soc. 1916, 38, 762). In this paper, Lewis proposed a general theory of bonding based on pairs of electrons shared between atoms. Lewis' ideas, coupled with the advent of quantum mechanics in the 1920s, led to the valence bond and molecular orbital theories, which remain the basis of most of today's bonding theory and computational chemistry.
In the foreword to the JCC special issue, Frenking and Shaik note that, even though there's plenty of work for historians to do in tracing the roots and development of chemical bonding models, "the field is exploding with new problems to be solved." The package of essays and papers demonstrates that "we are still far from understanding the nature of the chemical bond."

IN ONE EXPLORATION of virgin bonding territory, Hoffmann and former postdoc Dean J. Tantillo, who is now at the University of California, Davis, considered a new possibility for sigmatropic reactions by designing "sigmatropic shiftamers" (Acc. Chem. Res. 2006, 39, 477). Sigmatropic reactions are a type of rearrangement that can take place in hydrocarbons containing double bonds. The net effect is that σ bonds and π bonds trade places, which allows a hydrogen atom (or a double bond) to shift to a different part of the molecule. "Shiftamers are envisioned as polymers in which σ or π bonds migrate back and forth along the hydrocarbon framework, providing an unusual means of transporting electrons over long distances," Tantillo says.
Hoffmann and Tantillo began their investigation with polyacetylene, a conductive polymer in which the conduction of electrons or holes (missing electrons, or positive-charge carriers) arises from defects in the oxidized conjugated polymer structure. The scientists wondered if they could make analogs of conducting polymers by creating molecules in which sequential sigmatropic shifts swiftly unfold. They came up with two designs, which they call snakes and ladders.
The snakes are linear conjugated hydrocarbon chains that curl into a helix; sigmatropic shifts could occur for each section of the chain where the helix turns back on itself. The ladders are ladderanes, a pair of parallel hydrocarbon chains cross-linked by C-C bonds and adorned with double bonds that could participate in sigmatropic shifts. The shifts would allow the double bonds to migrate up and down the parallel chains. The researchers now await synthetic chemists to prove them right or wrong.
In another foray into new bonding territory, chemistry professor Kendall N. Houk of UCLA has been working to help refine the understanding of how enzymes work to accelerate reactions, which has been hard to pin down. By examining the binding strength of protein-ligand interactions in transition states, his group has shown that enzymes that accelerate reactions by a factor of more than 1011 must have a strong covalent or partially covalent bonding interaction with reacting substrates, in addition to any noncovalent interactions (C&EN, May 16, 2005, page 35). This controversial proposal is counter to the general emphasis in the field that transition-state interactions of the enzyme active site occur via noncovalent electrostatic interactions, hydrogen bonding, and hydrophobic interactions, Houk says.
Frenking's group, which is focused on developing a fundamental understanding of covalent bonding, recently has taken a new look at the possible valence states of carbon in organic compounds. Most synthetic organic chemistry involves tetravalent carbon compounds in which all four valence electrons partake in bonding. Stable divalent carbon compounds, such as singlet carbenes, which have a lone electron pair on carbon along with two substituents, are also well-known. An important example is N-heterocyclic carbenes, which have become useful ligands in transition-metal complexes and as organic catalysts.
Frenking and his coworkers have now presented theoretical and experimental evidence that formally zerovalent carbon can exist in compounds in which the valence electrons remain as two lone pairs that are not engaged in bonding (Angew. Chem. Int. Ed. 2006, 45, 8038). For this investigation, the researchers studied carbodiphosphoranes, such as C(PPh3)2, where Ph signifies a phenyl group. In reality, the carbon atom is divalent like in a carbene, but the electron pairs for the carbon-phosphorus bonds are donated entirely by the phosphorus atoms. This generosity leaves carbon with two lone pairs, and the carbon atom has a formal valence state of zero. Frenking and his coworkers report that this bonding picture is supported by the observed structure and high proton affinity of carbodiphosphoranes, as well as by the group's bond calculations.
For many inorganic and organometallic chemists, multiple bonding in the heavier main-group elements and in transition metals has become a hot topic. But it has been a long time in coming. Until the mid-1960s, chemists largely assumed that the triple bond was the highest possible multiple bond. In 1964, however, F. Albert Cotton and his coworkers at Texas A&M University surprised the chemistry community with evidence that the [Re2Cl8]2- ion contained a quadruple bond between two metal atoms, something that had never been seen before. Since then, chemists have observed many quadruple-bonded transition-metal compounds.

IT TOOK ANOTHER 40 years, but in 2005, Philip P. Power of UC Davis and his coworkers pushed the multiple-bond boundaries even further when they reported the first quintuple bond in the dichromium complex RCrCrR, where R designates a bulky terphenyl ligand (C&EN, Sept. 26, 2005, page 9). The researchers suggested that the two chromium(I) atoms, which have a 3d5 electron configuration, share five electron pairs in five bonding molecular orbitals. For his part, Power has been cautious about using the word "quintuple" to describe the novel bonding he and colleagues found. He prefers to call it "fivefold bonding" because the two metal ligands are in a "trans-bent" arrangement rather than in the linear arrangement expected for a traditional multiply bonded compound, which leads to an actual bond order of less than 5.0.
After the quintuple bond was announced, Power and his UC Davis colleague Marcin Brynda, along with Björn O. Roos and Per-Olof Widmark of Lund University, in Sweden, and Laura Gagliardi of the University of Geneva, in Switzerland, conducted a more extensive computational analysis of the dichromium compound (Angew. Chem. Int. Ed. 2006, 45, 3804). That study, which used a simpler PhCrCrPh model, supports the claim for a quintuple bond in the dichromium compound and its measured bond lengths. Clark R. Landis and Frank Weinhold of the University of Wisconsin, Madison, subsequently reported a theoretical study showing that the trans-bent geometry emerges from the s and d orbitals and how this geometry leads to the strongest possible bonding (J. Am. Chem. Soc. 2006, 128, 7335).
Roos and Gagliardi, along with Antonio C. Borin of the University of São Paulo, in Brazil, now report the results of an even broader computational study to show that the maximum multiplicity of a covalent chemical bond between any two elements is six and that the tungsten dimer (W2) has the distinction of being most likely to have the strongest sextuple bond (Angew. Chem. Int. Ed., DOI: 10.1002/anie.200603600). Some chemists had already recognized that in transition metals the s and d orbitals could combine to support a sextuple bond in a naked diatomic molecule—a compound in which there are no ligands to tie up a bonding orbital. A few sextuple-bonded compounds with a very short bond length, such as Cr2, have been trapped in the gas phase at low temperature and observed by laser-induced fluorescence spectroscopy.
It turns out the number of available valence electrons and available orbitals leads to a maximum bond order of 6.0 for only a few elements—the group 6 transition metals (chromium, molybdenum, and tungsten) and uranium. Among sextuple-bonded dimers made from these elements, Mo2 and W2 have the highest calculated actual bond order of 5.2. The W2 dimer claims the highest bond energy of the calculated sextuple bonds. The researchers attribute the higher energy to decreased electron repulsion in the 5d orbitals of tungsten, which are more diffuse than the 3d and 4d orbitals of chromium and molybdenum.
ACHIEVING EVEN higher multiple bonding would have to involve additional shells of atomic orbitals, such as f orbitals, the researchers point out. But their calculations show that the additional multiple bonds aren't likely to form because of the contracted nature of f orbitals. For example, a sextuple bond in U2, which calculations indicate is possible, has a lower bond energy because of participation of the 5f orbitals.
Even as some researchers succeed at gaining credibility for novel bonding concepts, such as higher-order bonding, others are developing unconventional bonding ideas that have yet to take hold. For example, Shaik's group, in collaboration with Philippe C. Hiberty's group at the University of Paris South, has been defining a class of electron-pair bonds called "charge-shift bonds" (Chem. Eur. J. 2005, 11, 6358).
Charge-shift bonding is not the result of traditional covalent or ionic bonding, but rather it arises from the resonance between the two, Shaik explains. For example, when the bonding is homonuclear, as between two fluorine atoms in F2, the covalent electron sharing is repulsive and the ionic component is overall nonpolar (neutral). There is instead a large stabilizing resonance energy generated by fluctuations in the electron density between the two atoms. This resonance energy overrides the repulsive covalency and provides a small net bond energy that holds the atoms together.
Advertisement
Besides occurring in homonuclear diatomic molecules such as F2, O2, and N2, the researchers predict, charge-shift bonding should show up in polar C-F bonds and in bonds of noble-gas compounds such as XeF2. It's also indicated as a feature in double and triple bonding and in most transition-metal complexes.
Charge-shift bonding appears to be common, Shaik says. But acceptance of the idea "would mean revising the electron-pair bond concept," he notes. Before the chemistry community would consider an extraordinary step like that, he adds, it will take independent theoretical derivations of charge-shift bonding and, more important, experimental observations by other groups. Shaik, Hiberty, and their coworkers, along with Wei Wu of Xiamen University, in China, believe they have identified the first potentially measurable example of the charge-shift resonance bond energy in competing hydrogen- and halogen-exchange reactions involving HF (J. Am. Chem. Soc. 2006, 128, 2836).
Shaik also has been involved in developing a model to explain the bonding situation in symmetrical cagelike metal clusters in which the metal atoms are strongly held together even though they do not share electron pairs (J. Phys. Chem. A 2006, 110, 8510). These "no-pair bonds" have a formal bond order of zero and occur when each atom has a single valence electron of the same spin. Examples of these no-pair bond clusters, such as Lin and Cun, have been explored computationally by Shaik's group and others.
Unpaired electrons with identical spins are unable to form a bonding pair because their spin states are required to be opposite of one another. But the cluster atoms are still weakly held together by what Shaik calls "ferromagnetic bonding," which he says is based on a covalent-ionic resonance interaction of the coupled electrons in the two atoms (a triplet-state electron pair).
The stabilizing effect of ferromagnetic bonding is delocalized and spread over the close neighbor atoms in a cluster. In effect, the electronic structure is a resonance hybrid of all the overlapping ferromagnetic bonds and is strong enough to stabilize the cluster relative to the energy of the dissociated (unbound) atoms. As a cluster gets larger and more unpaired electrons participate, the binding energy increases. This type of bonding shows how a weak interaction can become a "remarkably strong binding force, even without sharing an electron pair," Shaik says.
Shaik, working with his Hebrew University colleague David Danovich and postdocs Sam P. de Visser and Devesh Kumar, have calculated the bonding for clusters as large as Li12 and Cu14. The ferromagnetic binding energy reaches a plateau of about 19 kcal/mol per atom for Cu10 and above. At those binding energies, "it's clear we're not dealing with weak van der Waals interactions," Shaik notes. Signs of no-pair bonding have been observed spectroscopically in alkali-metal dimers, he says, and the bonds should be possible for other metals with single unpaired electrons in their valence shells.
The theoretical and computational toolbox for searching and analyzing novel bonding models only continues to grow. One of the more prominent models is the atoms-in-molecules (AIM) theory, an electron-density approach that Richard F. W. Bader of McMaster University, in Ontario, has been developing.
AIM depicts matter as pointlike nuclei embedded in an electron-density cloud. The molecular nature of such a system is based on the three-dimensional topology of the overall electron density and polarization of the density by electrostatic forces between nuclei and electrons. Because individual atoms or functional groups contribute to the overall properties of the molecule they form into, it's possible to deconstruct a molecule and do manageable calculations on the pieces with AIM theory. One can then add the properties of the pieces together to reconstruct the molecule.
AIM theory doesn't define a "bond," Bader says, because bonds don't exist. AIM theory instead indicates that "two atoms are bonded, as in a verb, not a noun." Bonded atoms are signified by a line, called the bond path, drawn between two atoms where the electron density is greatest, he says.
With AIM theory, chemists can predict any measurable physical parameter in a molecule, such as atomic charge, dipole moment, and bonding energies, Bader says. The presence of a bond path in any structure "unequivocally identifies the atoms as being bonded to one another, regardless of any assumed mechanism of bonding." Bader, who trained as a physical organic chemist, argues that a full understanding of bonding can only emerge from this firm physics perspective.
AIM is considered by some computational chemists to be one of the most important new bonding theories in recent decades. The theory is increasingly being applied to study an array of chemical systems, including solid-state materials and biomolecules. "AIM offers explanations for real problems because it deals with reality and physics," Bader says.
But Bader's matter-of-fact approach is not readily accepted by all computational chemists. Several researchers have found fault with the theory because it can lead to interpretations that give improbable results.
For example, AIM predicts that there is a bond path between two nearby helium atoms to form He2, similar to H2. Chemists accept that the binding interaction in H2 constitutes a bond, Frenking says. Although He2 has been spectroscopically observed at very low temperatures, the He-He attraction is so weak that it isn't considered a chemical bond. Chemists make a distinction between He2 and H2, while AIM theory puts them on equal footing, Frenking adds.
Frenking says he appreciates Bader for being the "physics conscience" of chemistry, because Bader "shows that physical laws may not be forgotten when we use chemical bonding models." AIM is an important contribution, Frenking believes, "but the theory has to be used with chemical sense and not just with physical rigor. What really matters is how theoretical methods and bonding models help to understand the material world on a molecular scale."
Part of Frenking's reasoning comes from his computational work to determine if there can be a bonding interaction between noble-gas atoms, as AIM predicts. His group has studied pairs of noble-gas atoms trapped inside a fullerene cage, such as Xe2@C60. Calculations indicate that Xe2@C60 is energetically favorable and has a short Xe-Xe separation, he tells C&EN. "But is this a bond?" he asks.
According to the definition of a chemical bond prescribed by the International Union of Pure & Applied Chemistry, it indeed is a bond, he says. According to IUPAC, a bond exists between two atoms or groups of atoms when the forces acting between them lead to an aggregation with sufficient stability to be considered an independent molecular species. "Xe2@C60 is, in principle, an observable species, which should be long-lived since the activation barrier for breaking it apart is quite high," Frenking says. "If it doesn't have that chemical bond, then what is Xe2@C60? It definitely is a molecule."
"The chemical community accepts the idea that the chemical bond can only be correctly understood today in the framework of quantum mechanics," Frenking and Shaik write in JCC. They support the "polyphonic culture" of computational chemistry that includes many different bonding models, and they wouldn't suggest trying to reduce this culture to a monolithic one with a single, dominant theory. "It would undermine the intellectual heritage of chemistry and its creative nature," Shaik says.
"From a distance of 90 years, we recognize that some of the reasoning applied by Lewis was wrong," Frenking and Shaik point out in the JCC special issue. "But despite all the modifications and qualifiers, the work of Lewis has had a tremendous impact on the evolution of chemistry, wherein the electron-pair bond concept remains a central guiding theme."
- History of Chemistry
- Chemical Bonding's Venerable Past
You might also like...


The power is now in your (nitrile gloved) hands
Sign up for a free account to get more articles. Or choose the ACS option that’s right for you.
Already have an ACS ID? Log in
Join the conversation
Contact the reporter
Submit a Letter to the Editor for publication
Engage with us on Twitter