Advertisement
Grab your lab coat. Let's get started
Welcome!
Welcome!
Create an account below to get 6 C&EN articles per month, receive newsletters and more - all free.
It seems this is your first time logging in online. Please enter the following information to continue.
As an ACS member you automatically get access to this site. All we need is few more details to create your reading experience.
Not you? Sign in with a different account.
Not you? Sign in with a different account.
ERROR 1
ERROR 1
ERROR 2
ERROR 2
ERROR 2
ERROR 2
ERROR 2
Password and Confirm password must match.
If you have an ACS member number, please enter it here so we can link this account to your membership. (optional)
ERROR 2
ACS values your privacy. By submitting your information, you are gaining access to C&EN and subscribing to our weekly newsletter. We use the information you provide to make your reading experience better, and we will never sell your data to third party members.
Pharmaceuticals
Big Hopes Ride On Big Rings
ACS Meeting News: Constraining molecules in macrocyclic rings could help address challenges in drug discovery
by Carmen Drahl
September 7, 2009
| A version of this story appeared in
Volume 87, Issue 36
A 15-membered ring might seem like too daunting a molecule to test in a drug discovery program. But in some cases, it might be just what the doctor ordered. At a symposium at last month’s ACS national meeting in Washington, D.C., researchers from industry and academe shared their experiences with macrocyclic rings before the Division of Medicinal Chemistry.
“As a tool in the drug discovery field, macrocycles are underutilized,” symposium organizer Paul M. Scola said in a phone interview. A senior principal scientist at Bristol-Myers Squibb (BMS), Scola noted that when applied strategically, a macrocycle—a compound with a ring of 12 or more atoms—may enhance binding affinity, target selectivity, and metabolic stability compared with its open-chain counterpart. Despite often defying Christopher Lipinski’s “Rule of Five,” a series of guidelines for determining whether a molecule is likely to be an orally active drug, macrocycles can possess druglike properties, he added.
That problem-solving potential notwithstanding, macrocycles represent just a small fraction of the total population of marketed drugs. Nearly all of those come from nature, with the antibiotic erythromycin and the immunosuppressant rapamycin as two examples. Because existing macrocyclic drugs are useful against infections, cancer, and other indications, chemists are trying to learn more about when a synthetic macrocycle might be a good choice. They are taking advantage of versatile reactions, such as olefin metathesis and click chemistry, to make more diverse macrocycles than ever before (Nat. Rev. Drug Disc. 2008, 7, 608).
Take Ensemble Discovery, a Cambridge, Mass.-based biotech company. There, chemists use DNA to guide the synthesis of thousands of different macrocycles in a single reaction vessel and then test their ability to disrupt biologically relevant interactions between proteins. The technology is built on work carried out in company founder David R. Liu’s lab at Harvard University. If a hit shows promise in initial tests, Ensemble can scale up that macrocycle’s synthesis without using DNA. “It’s perceived that macrocycles are tough to make, but we’ve solved all the real challenges so far,” Ensemble’s Chief Scientific Officer Nicholas K. Terrett said at the meeting.
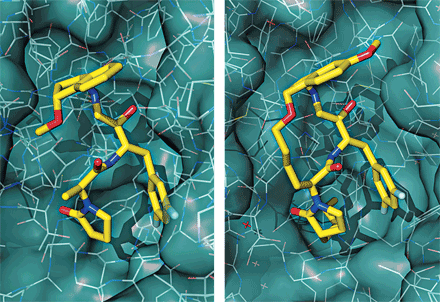
View Enlarged Image Most drugs target enzymes with well-defined pockets (left). Interactions between proteins (right) might be engaged by macrocycles.

View Enlarged Image Most drugs target enzymes with well-defined pockets (left). Interactions between proteins (right) might be engaged by macrocycles.
With this platform, Ensemble has discovered macrocycles that block the interaction of tumor necrosis factor (TNF) with its receptor, a notoriously tough-to-target interaction that underpins inflammatory diseases such as rheumatoid arthritis. So far, the only marketed drugs that combat this interaction are biologicals that must be injected. Terrett’s coworkers have found an orally active macrocycle called E-32712 that, via the TNF mechanism, is effective at treating rats with arthritic joints. The compound’s structure was not disclosed, but Terrett noted that it has been selected for further preclinical studies.
The potential to tune protein-protein interactions is a big reason why macrocycles are attracting attention, chemical biologist Paramjit S. Arora of New York University commented at the meeting. The vast majority of today’s drugs target enzymes, which usually have well-defined binding pockets. Interactions between proteins are harder to target because of the larger, flatter surface area involved.
“You typically can’t make compounds the size of [acetylsalicylic acid] for targeting a protein-protein interaction. That’s why we’re seeing molecules larger than that in this symposium,” Arora said.
Arora builds 13-membered rings to stabilize short peptides in α-helical conformations. Helices play key roles in many protein-protein interactions, including some implicated in HIV and cancer. By mimicking helical secondary structure, Arora hopes to build tools for probing protein-protein interactions, and perhaps even new therapeutics, he said at the symposium. To that end, his team recently described a set of computational tools for predicting which protein-protein interactions might be good targets for helix mimics (Mol. Biosyst., DOI: 10.1039/b903202a).
A portion of each macrocycle Arora uses acts as a surrogate for a hydrogen bond in a nascent helix. That molecular nudge is enough to encourage floppy, unstructured peptides, which ordinarily would never stably remain in a secondary structure, to coil into a helical shape on their own. Stabilized helices are more resistant to degradation by proteases and potentially enter cells more readily than linear peptides. Those benefits often hold true when drugmakers make macrocyclic drug candidates containing peptidic moieties.
Selectivity can be another of macrocycles’ perks, as Adam Nelson and coworkers at the University of Leeds, in England, learned. They modified the structure of a broad-spectrum kinase inhibitor, staurosporine, to make a family of macrocyclic analogs. “We were interested in the relationship of the macrocycle’s conformation to the molecule’s selectivity,” Nelson said at the meeting. Varying the macrocycle’s size altered its conformation and led to selective kinase inhibitors, which his team used to clarify the biological role of a particular kinase (Chem. Biol. 2009, 16, 15).
Meanwhile, at BMS, making a macrocycle helped chemists streamline a drug discovery program’s lead structure while also improving binding affinity for its target. At the session, senior research investigator Lawrence R. Marcin described a series of diaminopropanes that blocked the enzyme β-secretase, which is thought to be a desirable target for Alzheimer’s disease. Those molecules never reached clinical trials because they couldn’t penetrate brain tissues efficiently. While the BMS team pondered further adjustments, a molecular model of one compound bound to β-secretase provided a clue as to what to try next. “We saw that two portions of the molecule were intrinsically coming together on binding, and we realized we could tie them together as a macrocycle,” Marcin told C&EN.
To make macrocycles, the BMS team relied on a late-stage olefin metathesis reaction. Constraining their candidates in a ring gave a modest 10- to 20-fold enhancement in binding affinity for β-secretase and allowed the researchers to remove portions of the molecule that weren’t critical for activity. Unfortunately, the new candidates couldn’t reach the brain, either.

Macrocyclization often improves binding affinity, but it’s important to be careful when interpreting those benefits in thermodynamic terms, organic chemist Stephen F. Martin of the University of Texas, Austin, who did not attend the symposium, told C&EN. The prevailing belief is that preorganizing a molecule lowers the entropic penalty experienced on binding to a protein. But independent work by Martin and Mark R. Spaller of Dartmouth Medical School, in Hanover, N.H., suggests that’s not always true and that other factors may contribute to the boost in binding affinity (Angew. Chem. Int. Ed., 2006, 45, 6830; Biopolymers 2008, 89, 653). “The question of why this happens is something we’re still trying to sort out,” Martin said.
Macrocyclizations can be powerful tools for drug discovery, but the precise origins of enhanced binding affinities may vary with the system, Martin emphasized. To understand those reasons, it’s important to accurately measure enthalpies and entropies of binding with a technique such as isothermal titration calorimetry, he added.
Whatever its origin, the modest binding-affinity boost might still open up other options for the BMS team, Marcin noted. “A jump in potency lets you make changes elsewhere,” he explained. Make the right changes, and you could end up with a compound that goes all the way to clinical trials.
That’s what happened at Tranzyme Pharma, a biopharmaceutical company based in Research Triangle Park, N.C. Tweaking the tether component in a macrocyclic compound helped Tranzyme find its most advanced drug candidate, a therapy designed to get patients’ bowels moving again after surgery. The molecule, called TZP-101, has completed Phase IIb clinical trials.
Delays in recuperating gastrointestinal function, called postoperative ileus, are leading causes of extended hospital stays, Mark L. Peterson, vice president of intellectual property and operations at Tranzyme, said at the session. Because signaling by the hormone ghrelin boosts motility in the GI tract, Tranzyme sought a drug that would mimic that signaling action for treating postoperative ileus.

{kind=link}
Tranzyme used its proprietary MAcrocylic Template CHemistry (MATCH) technology to find drug candidates. In each of Tranzyme’s compounds, a short tether links the two ends of a tripeptide together, giving 14- to 20-membered rings. Small adjustments to that tether, such as removal of an olefin and addition of a methyl substituent with specific stereochemistry, were crucial for improving compounds’ solubility, half-life in blood plasma, and more, Peterson, an organic chemist, explained at the meeting.
TZP-101 is given intravenously, but further modifications have led to a variant that can be taken orally. The structure of that candidate, called TZP-102, was not disclosed.
Both molecules are also being evaluated as potential treatments for gastroparesis, a delayed emptying of food from the stomach that can be a complication of diabetes but has no known cause in 33% of cases. Current gastroparesis drugs are only moderately effective and have neurological or other side effects, so TZP-101 and 102 have both been granted fast-track status by the Food & Drug Administration, Peterson said.
Macrocycles aren’t a panacea, but their potential benefits should encourage medicinal chemists to consider them more often, Peterson told C&EN. “It’s time for macrocycles to become more visible in drug discovery. More people should know about the power of this structural class.”
The power is now in your (nitrile gloved) hands
Sign up for a free account to get more articles. Or choose the ACS option that’s right for you.
Already have an ACS ID? Log in
Join the conversation
Contact the reporter
Submit a Letter to the Editor for publication
Engage with us on Twitter