Advertisement
Grab your lab coat. Let's get started
Welcome!
Welcome!
Create an account below to get 6 C&EN articles per month, receive newsletters and more - all free.
It seems this is your first time logging in online. Please enter the following information to continue.
As an ACS member you automatically get access to this site. All we need is few more details to create your reading experience.
Not you? Sign in with a different account.
Not you? Sign in with a different account.
ERROR 1
ERROR 1
ERROR 2
ERROR 2
ERROR 2
ERROR 2
ERROR 2
Password and Confirm password must match.
If you have an ACS member number, please enter it here so we can link this account to your membership. (optional)
ERROR 2
ACS values your privacy. By submitting your information, you are gaining access to C&EN and subscribing to our weekly newsletter. We use the information you provide to make your reading experience better, and we will never sell your data to third party members.
Synthesis
Science Concentrates
May 2, 2005
| A version of this story appeared in
Volume 83, Issue 18
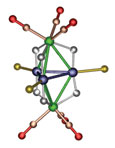
A platinum-rhenium bimetallic cluster with an unusually high degree of electronic unsaturation around the metal atoms has been discovered to add an equally unusual amount of hydrogen, forming a hydride complex. The discovery could provide new insight into hydrogenation catalysis and hydrogen storage applications, according to Richard D. Adams and Burjor Captain of the University of South Carolina, Columbia, who conducted the research (Angew. Chem. Int. Ed. 2005, 44, 2531). Adams' group has been synthesizing platinum and palladium compounds containing the bulky tri-tert-butylphosphine ligand (L), including a trigonal bipyramid cluster, Pt3Re2(CO)6(L)3, which is deficient by 10 electrons. It reacts at room temperature with H2 (a good source of electrons) to form the hexahydride complex shown (Re is green; Pt, purple; H, white; P, yellow; tert-butyl groups omitted; and CO, red). "The addition of three equivalents of H2 to an intact cluster complex is unprecedented," Adams says, "although there are examples of complexes that contain more hydrogen than this one."
Two ligands play independent roles in bringing about different steps of a catalytic cycle, scientists have observed. The phenomenon, dubbed serial ligand catalysis, may be occurring widely, but the work of M. Christina White and coworkers at Harvard University may be the first to explicitly demonstrate it (J. Am. Chem. Soc., published April 21, dx.doi.org/10.1021/ja0500198). They obtained evidence for serial ligand catalysis from a mild and highly selective palladium-catalyzed allylic oxidation of terminal olefins. The data are consistent with a palladium-phenyl vinyl sulfoxide complex promoting the initial cleavage of the allylic carbon-hydrogen bond followed by a palladium-benzoquinone complex promoting the functionalization of the allylic carbon. The conventional approach to homogeneous catalysis of one-metal/one-ligand combination may be inefficient when a reaction involves several product-forming steps that impose different demands on the metal, White says. "One-metal/multiple-ligand combinations may result in uniquely mild and selective solutions."

A prototype microbial fuel cell designed to generate a steady stream of electricity as it cleans wastewater now has been modified to produce hydrogen instead (Environ. Sci. Technol., published April 22, dx.doi.org/10.1021/es050244p). Bruce E. Logan and Hong Liu at Penn State University originally designed their flow-through fuel cell to use bacteria living on the carbon anode to oxidize organic matter in wastewater. Hydrogen ions and electrons generated by the oxidation combine at the cathode with O2 from air to form water and generate electricity. Bacteria have a "fermentation barrier" that limits their ability to completely degrade carbohydrates to CO2 and H2, but the Penn State researchers determined that excluding O2 from the system and applying an additional 0.25 V to the circuit could overcome the barrier. The modified fuel cell efficiently generates H2 from acetic acid in the lab, but any type of organic matter in wastewater would work. The researchers believe their fuel cell could supply enough H2 for energy production to significantly offset the cost of wastewater treatment.

An exotic "proton sandwich" cation may be the key intermediate in some terpenoid cyclizations, according to theoretical chemists at the University of California, Davis. In computational studies of terpenoid cyclization mechanisms, Dean J. Tantillo and Pradeep Gutta encountered an unusual nonclassical cation with a five-center, four-electron bonding motif (Angew. Chem. Int. Ed. 2005, 44, 2719). In the sandwichlike carbocation, "a formally tetracoordinate proton is seemingly suspended between two C=C bonds on opposite sides of a cyclooctadiene ring," Tantillo and Gutta note. Further calculations on a simplified 1,5-cyclooctadiene system show that the proton is shared equally by all four "alkene" carbons (shown) with C...H bond lengths of 1.48 Å or 1.46 Å , depending on the parameters of the calculation. These distances are longer than those in both cyclic and acyclic three-center, two-electron C...H...C cations, which typically have bond lengths around 1.3 Å. The authors are currently studying the cation as a possible intermediate in the biosynthetic pathway between the farnesyl cation and pentalenene, a tricyclic terpenoid natural product.
Advertisement
Transfer of chirality from DNA to the products of a chemical reaction has been shown (Angew. Chem. Int. Ed., published April 21, dx.doi.org/10.1002/anie.200500298). The demonstration may lead to a new area of catalyst design, according to Gerard Roelfes and Ben L. Feringa of the University of Groningen, in the Netherlands. They anchored a copper(II)/ligand complex to DNA through a well-known DNA intercalator. Although the ligand is nonchiral, when the catalytic system is applied to an asymmetric Diels-Alder reaction in water, the products are formed in significant enantiomeric excess. Furthermore, either enantiomer of the Diels-Alder products can be formed in significant excess by adjusting the distance of the ligand from the DNA. DNA presents numerous opportunities for catalyst design. For example, the DNA sequence can be tailored to bind different intercalators bearing different catalytic systems at predetermined positions. DNA could be "an important scaffold for the design and discovery of new catalytic systems for a wide range of reactions and maybe even multistep conversions," Roelfes says.
The power is now in your (nitrile gloved) hands
Sign up for a free account to get more articles. Or choose the ACS option that’s right for you.
Already have an ACS ID? Log in
Join the conversation
Contact the reporter
Submit a Letter to the Editor for publication
Engage with us on Twitter