Advertisement
Grab your lab coat. Let's get started
Welcome!
Welcome!
Create an account below to get 6 C&EN articles per month, receive newsletters and more - all free.
It seems this is your first time logging in online. Please enter the following information to continue.
As an ACS member you automatically get access to this site. All we need is few more details to create your reading experience.
Not you? Sign in with a different account.
Not you? Sign in with a different account.
ERROR 1
ERROR 1
ERROR 2
ERROR 2
ERROR 2
ERROR 2
ERROR 2
Password and Confirm password must match.
If you have an ACS member number, please enter it here so we can link this account to your membership. (optional)
ERROR 2
ACS values your privacy. By submitting your information, you are gaining access to C&EN and subscribing to our weekly newsletter. We use the information you provide to make your reading experience better, and we will never sell your data to third party members.
Pharmaceuticals
Drugs from Academia
Some drug discovery efforts are anything but 'academic'
by Stu Borman
April 16, 2007
| A version of this story appeared in
Volume 85, Issue 16
CHEMIST DENNIS C. LIOTTA of Emory University enjoys citing dictionary definitions of the two words in the phrase "academic research." In his presentation to a Division of Medicinal Chemistry session on marketed drugs discovered in academia at last month's American Chemical Society national meeting in Chicago, Liotta noted that "research" means a "careful or diligent search" or a "studious inquiry or examination." "It's a very nice, noble kind of activity," he added.
"Academic" is a horse of a different color. Liotta pointed out that in its noun form, it's defined as a person who is "very learned but inexperienced in practical matters." In its adjectival form, he continued, its definitions include "theoretical without having an immediate or practical bearing" and "having no practical or useful significance." One of the word's synonyms is "pedantic," Liotta added.
"Sometimes when we call something 'academic,' that means it doesn't matter," Liotta said. "I want to do things that matter. I certainly don't want to be associated with terms like 'pedantic.' "
Liotta's research achievements have not been merely academic, in the pejorative sense of that term. After all, he and coworkers discovered a couple of the most important anti-HIV drugs now on the market.
In fact, all five speakers at the ACS session devised successful drugs (or, in one case, a key drug production method) in their academic laboratories. All of the drugs with which they're associated have been remarkably successful, and the five speakers have presumably all done well financially as a result of those successes.
The drug that chemist Ronald Breslow of Columbia University invented with cell biologist Paul A. Marks and coworkers at Memorial Sloan-Kettering Cancer Center is the anticancer agent suberoylanilide hydroxamic acid (SAHA). In 1971, Breslow recounted, Charlotte Friend, a microbiologist and oncologist at Mount Sinai School of Medicine (who died of lymphoma in 1987), found that dimethyl sulfoxide (DMSO) caused cancerous red blood cells to lose their disease traits, thereby permitting them to differentiate into healthy erythrocytes. 'Twas a consummation devoutly to be wished, as Shakespeare might have put it—the seemingly miraculous transformation of a tumor cell into a normal one.
However, DMSO didn't exhibit a high level of this medicinal activity. So in 1974, Breslow, Marks, and coworkers set out to find structurally related compounds that might have anticancer effects more potent than those of DMSO. Although the researchers did not know DMSO's biological target, they still managed to surpass the potency of DMSO by a few orders of magnitude by making a series of well-chosen structural changes in the compound. One of the more potent analogs they developed was SAHA.
"That's the one we decided to pursue" as a potential therapeutic agent, Breslow said. "We didn't know how it worked. We didn't know what else it would do. But since it was doing wonderful things with erythroleukemia cells" and many other types of cancer cells in culture, Breslow noted, "we decided to run with it and see how far it went." Pharmaceutical companies would like to be that lucky a little more often, as SAHA is now a Food & Drug Administration-approved drug.
The biological target of SAHA was later found to be histone deacetylase, an enzyme that helps regulate gene transcription. In 1999, they, in collaboration with the group of structural biologist Nikola Pavletich (now at Memorial Sloan-Kettering Cancer Center), obtained the first crystal structure of SAHA bound to a histone deacetylase homolog.

ANALOGS OF SAHA, including some designed by Merck, also turned out to be highly potent against cancer cells. But the most potent ones were unfortunately quite toxic. "Merck concluded that this class of compounds could not succeed, and they closed the program," Breslow said. "It was a perfectly good conclusion, except that we had one that did work and could be used in tolerable doses," Breslow said, referring to SAHA. "High potency didn't work, but medium potency was still okay. The point of this game was to get a drug, not to set the world's record for highest potency."
In Phase I and II clinical trials for cancer, SAHA showed good oral availability and no serious toxicity, even at high doses, although fatigue and gastrointestinal side effects have been observed. Most of the patients showed "useful responses," Breslow said. The drug appeared to be most promising as a combination therapy with other anticancer compounds or with radiation.
"If you are lucky enough to get a drug that makes it through Phase I and II, your troubles have now started," Breslow said. In 2001, he and his colleagues founded a small company called Aton Pharma to develop SAHA and related agents. "We raised about $70 million from investors, and we were able to do the Phase I and II trials. But at the point where we got to Phase III, we decided we really couldn't do it. So what we did instead was to shop the compound around," he said.
Merck, which earlier had shut down its histone deacetylase inhibitor program, bought Aton in 2004 and put SAHA into Phase III trials. FDA granted SAHA fast-track and orphan-drug status because it seemed to represent a promising agent for cutaneous T-cell lymphoma patients who had not responded to existing therapies.
Merck completed its clinical trials, submitted the agent for approval in June 2006, and got FDA approval last October for SAHA, which it now markets as vorinostat (Zolinza).
Many additional clinical trials of SAHA—currently more than 40—are under way for conditions such as brain cancer, mesothelioma, kidney and breast cancer, lymphoma, leukemia, melanoma, and ovarian, thyroid, colorectal, metastatic, and non-small-cell lung cancers because the agent seems to have an extraordinarily broad range of activity. SAHA also has recently been shown to stop the progression and cause regression of Huntington's chorea in an animal model of the disease, Breslow said.
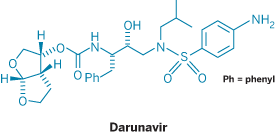
Other recently approved drugs have academic roots, too. If you go to the Web page of Arun K. Ghosh's medicinal chemistry group at Purdue University, you'll see the following notice: "Good News! The U.S. FDA has recently approved darunavir." Ghosh and coworkers take great pride in having discovered darunavir, a protease inhibitor designed to combat drug-resistant forms of HIV.
Saquinavir, the first approved protease inhibitor, came out in 1996, and several other protease inhibitors were subsequently developed and approved. Although HIV-infected patients initially respond well to most protease inhibitors, HIV tends to develop resistance to the agents, and the drugs tend to lose efficacy within six to eight months of the start of treatment.
In an effort that started strictly as an educational project in the mid-1990s, Ghosh and coworkers began investigating ways, in Ghosh's words, "to alleviate poor absorption properties of saquinavir-like molecules and specifically to design new protease inhibitors with improved activity against drug-resistant viral strains." Their idea was to develop compounds that would bind to the relatively invariant backbone structure of the HIV protease protein. Existing protease inhibitors, including saquinavir, primarily targeted the protease's amino acid sidechains, which the virus could modify more easily.
THEY USED structure-based drug design, based on X-ray crystallographic analysis of inhibitor-bound HIV protease structures, to optimize inhibitor binding to the enzyme backbone. This approach led to the creation of darunavir.
Ghosh and coworkers then began collaborating with Hiroaki Mitsuya, head of the Experimental Retrovirology Section at the National Cancer Institute (NCI), to evaluate darunavir's medicinal potential. Mitsuya's tests of the drug with live HIV variants in human cells showed that the agent maintained "incredible potency" against drug-resistant HIV, Ghosh said. In fact, he noted, its potency was better than that of all the other protease inhibitors with which it was compared.
The drug subsequently did well in clinical trials sponsored by Bridgewater, N.J.-based Tibotec Therapeutics. It received accelerated approval by FDA last June and is now marketed as Prezista. The protease inhibitor is approved for treatment of HIV and AIDS patients harboring multi-drug-resistant HIV variants that have not responded to existing combination drug regimens. "It is likely to have significant impact on the fight against AIDS," Ghosh said.
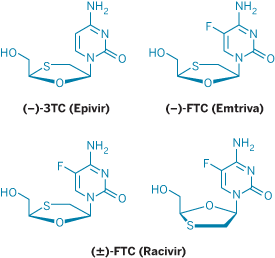
Meanwhile, Liotta and colleagues were discovering other important drugs in the anti-HIV armamentarium. Among them are 3TC or lamivudine (Epivir), marketed by GlaxoSmithKline and Shire Pharmaceuticals, and FTC or emtricitabine (Emtriva), marketed by Gilead Sciences. The researchers also discovered three other anti-HIV drugs that are currently in Phase II clinical trials.
FTC and 3TC are oxathiolane nucleoside analogs. The story of how these agents were developed goes back to the late 1980s, when the reverse transcriptase inhibitor zidovudine (Retrovir or AZT) became the first approved drug for treatment of HIV infections. "It gave miraculous results for about six months, and then the people who were taking it relapsed as a result of resistance," Liotta said.
At a 1989 AIDS conference, a racemic drug called BCH-189, which had been synthesized by Canadian researchers, was reported to show good activity against HIV and relatively low toxicity in cellular assays. Emory University professor of pediatrics and chemistry Raymond F. Schinazi asked Liotta if he could develop a synthesis for the agent. "I was reluctant to get involved," Liotta said, because he believed prior patent protection of the racemic compound would prevent its development by the Emory group. But he took on the project anyway.
Liotta and coworkers initially developed a BCH-189 synthesis that Liotta describes as "really primitive." But the team soon devised a more sophisticated version of the synthesis based on tin complexation of a key starting material. The researchers began using the improved method to create analogs, and they also developed a way to separate enantiomeric forms of the agents produced by the racemic procedure.
"Suddenly, we had an incredibly versatile method for making these compounds," Liotta said. "And we could run through a [structure-activity relationship study] very quickly, while our friends in industry who were trying to move these same compounds forward were slugging it out and having a very difficult time. Having the right chemistry gave us an opportunity to run circles around our competition." Glaxo later licensed the Emory process for making BCH-189.
By screening BCH-189 analogs for activity, the Emory researchers found two interesting anti-HIV agents. But in culture, HIV exposed to the compounds quickly developed a unique mutation close to the active site of its reverse transcriptase enzyme, boosting its resistance to the agents by more than 1,000-fold. "We figured that these compounds were dead," Liotta said. However, the 1,000-fold-resistant mutant turned out to be hypersensitive to AZT, giving it an Achilles' heel, and this observation gave the researchers the confidence to continue working on the agents.
One of these agents was 3TC, which was later approved as an anti-HIV therapeutic. "It proved the basis of the first rational combination therapy," Liotta said, referring to treatment based on 3TC and AZT. 3TC later became the first approved drug for treating hepatitis B infection as well.
For some time, 3TC was a subject of legal disputes between the Emory group and several pharmaceutical companies. "We didn't exactly see eye-to-eye over whose rights were prevalent," Liotta said, "so we had fairly protracted litigation. But later on we resolved all those issues."
Liotta, Schinazi, and coworkers also discovered FTC, a fluorinated version of 3TC. Triangle Pharmaceuticals (later acquired by Gilead Sciences) and Burroughs Wellcome put the drug through clinical trials. In 2003, FDA approved FTC for treatment of HIV infection in combination with other agents. FTC has very low toxicity and has become a major anti-HIV therapy.
Advertisement
"3TC and FTC together comprise about 40% of the nucleoside reverse transcriptase inhibitor market," Liotta said. "What that means is, since drugs are given in combination and nucleoside reverse transcriptase inhibitors are always present, just about everyone who has HIV has taken one of these drugs." Not bad for academic research.
3TC and FTC are enantiomeric drugs that are relatively expensive to produce. Over a decade ago, Schinazi suggested that racemic FTC (now trade named Racivir) might be a good candidate for development as well because it could be produced inexpensively and therefore might be particularly valuable for managing HIV infections in the developing world. At the time, Liotta said the idea would never work, but he helped develop FTC anyway, and lately his opinion about the drug's prospects has changed. "It looks like it might have some opportunities to be a salvage therapy for patients who have run out of options," Liotta said. It's currently in Phase II trials sponsored by Pharmasset, a Princeton, N.J.-based company cofounded by Schinazi and Liotta.
THE STORY of the drug discovered by professor of chemistry emeritus Edward C. Taylor of Princeton University goes further back. About 60 years ago, when Taylor was entering grad school at Cornell University, he was searching for an interesting thesis topic and happened to see an article in Science by researchers at Lederle Laboratories on pteroylglutamic acid, a factor isolated from human liver that appeared to be necessary for the growth of microorganisms. The agent, now called folic acid, contained a curious bicyclic heterocycle that had only been seen previously in pigments from the wings of butterflies. "I thought that a study of the chemistry and biological role of this and related ring systems would make a wonderful research project for my Ph.D. thesis," Taylor said.
Lederle researchers subsequently made some analogs of folic acid, including methotrexate and aminopterin. Aminopterin was too toxic to function as an antibacterial agent. In fact, its toxicity has been in the news lately because it has been used as a rat poison and was initially believed to be the toxic material in recalled pet food (C&EN, April 2, page 11). "If I'd known that, I might have given up trying to make useful drugs by small chemical modifications of folic acid, but I'm glad I didn't," Taylor said.
Methotrexate was not quite as toxic, and in the early 1950s, researchers discovered that it brought about remissions of acute lymphoblastic leukemia in children. It was an inhibitor of dihydrofolate reductase, a folate-dependent enzyme required for the biosynthesis of DNA and RNA.
In an effort to find inhibitors of other folate-dependent enzymes, Taylor and coworkers began synthesizing analogs of folic acid beginning in the 1970s. One of these analogs was a compound called 5,10-dideaza-5,6,7,8-tetrahydrofolic acid (DDATHF). Taylor's group, in collaboration with Eli Lilly & Co. researchers, found that DDATHF blocked purine biosynthesis and showed unusually powerful anticancer activity against an extraordinarily wide range of solid tumors, including some that had developed resistance to methotrexate.
Princeton and Lilly agreed to collaborate further to explore this discovery. With Lilly research support, Taylor and coworkers made a large number of structure-based design changes to solve some stereochemistry-related problems encountered with DDATHF. One of the resulting compounds eventually became the approved drug pemetrexed, which Lilly now markets as Alimta.

In early 2004, FDA approved a combination therapy of Alimta with cisplatin for treatment of malignant pleural mesothelioma. "It's the only drug that has been approved for this type of tumor," Taylor said. "It's now the world standard for treatment of malignant pleural mesothelioma." Alimta was also approved later that year as a single agent for treatment of non-small-cell lung cancer, and it is currently the leading treatment in the U.S. for refractory cases of this type of cancer, Taylor said.
Alimta's toxicity is readily manageable by coadministration of folic acid and vitamin B-12, Taylor said. What's more, he noted, every solid tumor examined so far in clinical trials has responded to it. It is currently in dozens of clinical trials, both in combination with other cancer drugs and as a single agent, for a wide range of solid tumors. "It's been on the market now only two full years, and Lilly reports that, based on sales, it is the most successful cytotoxic drug launch in history," Taylor said.
Lilly supported Taylor's academic research throughout the drug discovery process and was granted an exclusive license to market Alimta. The patent for Alimta is owned by Princeton, and Lilly pays the university royalties for the license. Princeton is currently using part of those royalties to build a new chemistry building.
Chemist Robert A. Holton of Florida State University (FSU) also played an important role in development of a major cancer drug. But when it was his chance to speak at the symposium, he said, "I'm probably the one who doesn't belong here." The reason? "I believe the title of this symposium is something like 'drugs developed in academic laboratories,' and we didn't develop a drug," he said. "But I have a story to tell anyway."
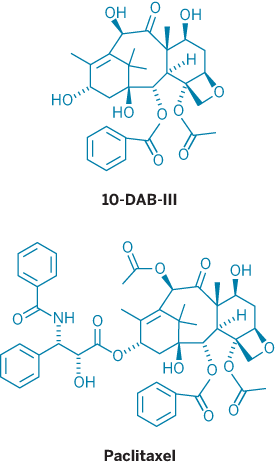
Holton recounted that around 1960, NCI and the Department of Agriculture started a joint program to screen compounds from plants to find new compounds of biomedical interest. One of the compounds found that way was the anticancer agent paclitaxel. Derived from the bark of yew trees, paclitaxel was found to be biologically active in initial screening tests. By the early 1970s, paclitaxel had been isolated and structurally analyzed by a group at Research Triangle Institute.
The group reported the structure in the Journal of the American Chemical Society. "That was where I first saw it, when I was a postdoc at Stanford," Holton said.
In 1979, biologist Susan Band Horwitz of Albert Einstein College of Medicine discovered that paclitaxel had a unique mechanism of action—stabilization of cell microtubules and consequent inhibition of cell division. This discovery raised the agent's profile, and NCI began Phase I clinical trials to test paclitaxel against several cancer types in 1984.
However, development of the drug could only proceed slowly "because the supply was pitiful," Holton said. It was very difficult to obtain from the yew bark in which it had been found. The yield of this process was about 0.004% and required killing the trees, Holton said. Holton and coworkers had already been interested in developing a total synthesis of paclitaxel, and the supply problem increased their resolve. Nevertheless, "at that time, I never believed that [paclitaxel] would ever be a drug," he said.
"In the mid-80s, I went to Susan's laboratory at Einstein and talked to her for quite a while," Holton recounted. "Susan said to me, 'I really admire what you natural products chemists do, but your research does not help me or cancer patients. You use many steps, you use intricate reactions, and in the end, you don't make enough of a compound to do anything with.' "
Holton and coworkers nevertheless continued to work on a total synthesis while paclitaxel was in clinical trials. "I didn't know much about the clinic. Didn't mean much to me at all," Holton said.
Responses of ovarian cancer and breast cancer patients to paclitaxel in the late 1980s proved to be "pretty phenomenal," Holton said. "This got everyone's attention." The late Matthew Suffness, who was program director for paclitaxel-related grants at NCI, "started calling everyone saying, 'Look, this is going to be a drug. We don't have any way to really get it. There's a huge supply problem. So all you people who are working on the total synthesis need to pay some attention to this.' "
One person who had been paying some attention was French natural products chemist Pierre Potier, who died last year. In the early 1980s, Potier and coworkers developed not a total synthesis but a more practical semisynthetic approach to the production of paclitaxel-like compounds. Potier's group found that a compound called 10-deacetylbaccatin-III could be extracted easily and in large quantities from the needles of Taxus baccata, the English yew. They also found that acetylating this compound and attaching a sidechain to it yielded much larger quantities of paclitaxel-like compounds than had been available before. Rh??ne-Poulenc Rohrer eventually made one of these analogs into the approved anticancer agent docetaxel (Taxotère).
In 1988, Potier, along with Andrew E. Greene of Joseph Fourier University, Grenoble, France, and coworkers developed an esterification technique for adding the sidechain to the paclitaxel ring system. But the yield was only fair, and Holton felt it could be improved considerably. So, he and his coworkers developed a more efficient semisynthesis based on a β-lactam reagent, and FSU applied for a patent on the process in June 1989.
SOON THEREAFTER, Bristol-Myers Squibb (BMS) formed an agreement with the National Institutes of Health to develop paclitaxel, and the company licensed Holton's semisynthetic process to produce the agent. The terms of the BMS-FSU license included a five-year grant from BMS to FSU of $1.7 million to support Holton's research on the synthesis and evaluation of paclitaxel analogs. "I felt this was a huge amount of money and that my ship had come in," Holton said.
Advertisement
In the early 1990s, he and his coworkers further improved the yield of the semisynthesis by using a lithium alkoxide reagent to add the paclitaxel sidechain to the ring system, and BMS licensed that process as well. BMS obtained FDA approval for paclitaxel on Dec. 29, 1992, and began marketing it as Taxol.
About a year later, Holton and coworkers finally completed a total synthesis of the compound. Chemistry professor K. C. Nicolaou and coworkers at Scripps Research Institute and the University of California, San Diego, also completed a total synthesis at about the same time. But the two total syntheses, like most total syntheses, were woefully inefficient.
"Remember when I told you that Susan said we don't ever make enough stuff to be of use to her?" Holton asked. "She was right." The total synthesis required over 40 reactions and gave a 2% yield, whereas the semisynthesis required only four reactions and gave an 80% yield. Although the race to achieve the first total synthesis got a lot of attention, the semisynthesis turned out to be far more practical, economically important, and lucrative.
Paclitaxel has been a very successful anticancer drug, and from 1993 until this month, FSU has received more than $350 million in royalties from Holton's semisyntheses. Holton noted that the FSU royalty distribution (after expenses), is a matter of state law: 40% to the inventor (Holton), 30% to the inventor's department, and 30% to the university.
Holton has been generous with his royalties. In 1995, he formed MDS Research Foundation, which supports basic research in molecular design and synthesis. MDS Research Foundation later established the SynCure Cancer Research Foundation, a charitable organization, and Taxolog, a company devoted to the development of paclitaxel analogs.
While paclitaxel was being marketed, Holton and coworkers, with BMS support, created over 500 analogs, some of which have better efficacy than the parent compound. In 1995, BMS concluded that the more efficacious analogs were too toxic, and it therefore returned rights to all the analogs to FSU. In 1998, Taxolog licensed these analogs from FSU and began creating thousands of others. Many of these analogs have been screened against human tumors, and some appear to work better than paclitaxel. At this point, Taxolog has three drugs in clinical trials—milataxel in Phase II and simotaxel and TL310 in Phase I. So the story of paclitaxel continues to unfold.
Holton noted that Gilbert Stork of Columbia University once said to him that, in the paclitaxel semisynthesis, "all you did was make an ester." Holton replied, "That's true, but it was a good ester to make."
You might also like...


The power is now in your (nitrile gloved) hands
Sign up for a free account to get more articles. Or choose the ACS option that’s right for you.
Already have an ACS ID? Log in
Join the conversation
Contact the reporter
Submit a Letter to the Editor for publication
Engage with us on Twitter