Advertisement
Grab your lab coat. Let's get started
Welcome!
Welcome!
Create an account below to get 6 C&EN articles per month, receive newsletters and more - all free.
It seems this is your first time logging in online. Please enter the following information to continue.
As an ACS member you automatically get access to this site. All we need is few more details to create your reading experience.
Not you? Sign in with a different account.
Not you? Sign in with a different account.
ERROR 1
ERROR 1
ERROR 2
ERROR 2
ERROR 2
ERROR 2
ERROR 2
Password and Confirm password must match.
If you have an ACS member number, please enter it here so we can link this account to your membership. (optional)
ERROR 2
ACS values your privacy. By submitting your information, you are gaining access to C&EN and subscribing to our weekly newsletter. We use the information you provide to make your reading experience better, and we will never sell your data to third party members.
Synthesis
Virtually Created Enzymes
Designed retro-aldolases catalyze breaking of carbon-carbon bond in an unnatural substrate
by Elizabeth K. Wilson
March 10, 2008
| A version of this story appeared in
Volume 86, Issue 10
IN A DRAMATIC EXAMPLE of translation from in silico to in vitro, scientists have computationally designed a series of new enzymes that catalyze a multistep reaction-including the breaking of a carbon-carbon bond-in laboratory experiments.
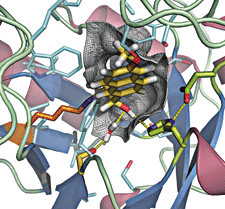
This computationally designed enzyme, whose catalytic center is shown here, catalyzes the retro-aldol reaction. The groups important for catalysis include the nucleophilic imine-forming lysine (orange) and hydrogen-bonding groups (light green).
The designed enzymes, which catalyze the reverse of an aldol condensation reaction, aren't nearly as active as naturally occurring enzymes that perform similar tasks (Science 2008, 319, 1387). But the fact that they work on a substrate not found in nature suggests that computer-aided design might yield enzymes that can perform nearly any reaction a chemist desires.
Biochemistry professor David Baker, graduate student Lin Jiang, and postdoc Eric A. Althoff of the University of Washington, Seattle, and their colleagues used the Rosetta protein structure prediction program developed in the Baker lab to design a library of enzymes with active sites likely to catalyze the so-called retro-aldol reaction. They whittled down a virtual list of more than 180,000 candidates to a promising 72.
The team then synthesized those enzyme candidates in the lab. Thirty-two enzymes catalyzed the reaction, earning them the name retro-aldolase; some accelerated the reaction rate by up to four orders of magnitude. Using X-ray crystallography, the researchers confirmed that the synthesized enzymes' structures matched those they had designed computationally.

"I think it's a real breakthrough for computational design," says Howard Hughes Medical Institute Investigator Jack W. Szostak of Massachusetts General Hospital, Boston. His lab has its own strategies for designing enzymes (C&EN, Aug. 20, 2007, page 13). "Even though there is still a long way to go to be able to design enzymes as good as those found in nature, this is a big step," he says.
Generally, retro-aldolases, which are biologically common, break β-hydroxycarbonyl compounds into two carbonyl derivatives. Instead, the group chose 4-hydroxy-4-(6-methoxy-2-naphthyl)-1-butanone as a substrate, which isn't found in biological systems.
Because the retro-aldol reaction involves a number of steps with different transition states, the group designed an active site for the enzyme that superimposed all of the transition states on top of each other. The design placed the catalytic residues optimally for all key steps and accounted for conformational changes as well, Jiang and Althoff explain.
The authors emphasize that although they have demonstrated their artificial enzymes have catalytic activity, comparable natural retro-aldolases catalyze their natural substrates about 100,000 times more efficiently.
The group hopes to generalize the information about activity gleaned from these experiments to their design process. The sheer number of active designs bodes well for the approach, Jiang and Althoff tell C&EN.
Advertisement
The power is now in your (nitrile gloved) hands
Sign up for a free account to get more articles. Or choose the ACS option that’s right for you.
Already have an ACS ID? Log in
Join the conversation
Contact the reporter
Submit a Letter to the Editor for publication
Engage with us on Twitter