Advertisement
Grab your lab coat. Let's get started
Welcome!
Welcome!
Create an account below to get 6 C&EN articles per month, receive newsletters and more - all free.
It seems this is your first time logging in online. Please enter the following information to continue.
As an ACS member you automatically get access to this site. All we need is few more details to create your reading experience.
Not you? Sign in with a different account.
Not you? Sign in with a different account.
ERROR 1
ERROR 1
ERROR 2
ERROR 2
ERROR 2
ERROR 2
ERROR 2
Password and Confirm password must match.
If you have an ACS member number, please enter it here so we can link this account to your membership. (optional)
ERROR 2
ACS values your privacy. By submitting your information, you are gaining access to C&EN and subscribing to our weekly newsletter. We use the information you provide to make your reading experience better, and we will never sell your data to third party members.
Pharmaceuticals
Cystic Fibrosis Letdown Explained
Drug Combo: Cell studies suggest way to boost clinical outcomes
by Carmen Drahl , Lisa M. Jarvis
July 24, 2014
| A version of this story appeared in
Volume 92, Issue 30

One of the two drugs in a promising cystic fibrosis therapy might be undoing the beneficial effects of the other, two studies suggest. The work, conducted in cells, may explain the modest results from a major clinical trial and provide an avenue for improved drugs.
People with cystic fibrosis endure lung-clogging mucus and debilitating infections. Until recently, there was no way to fix the underlying cause of the disease, a malfunctioning chloride ion channel protein called the cystic fibrosis transmembrane conductance regulator (CFTR).
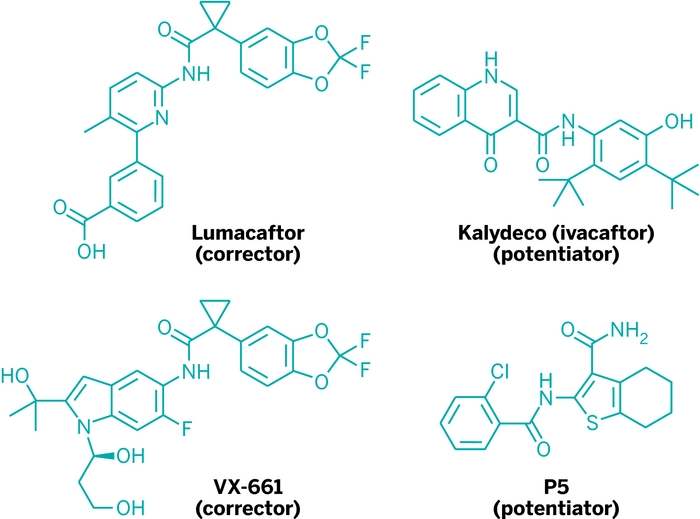
That changed with the 2012 debut of Kalydeco (ivacaftor). The drug boosts chloride ion flow through CFTR. Kalydeco works reasonably well on people with a rare gene mutation that locks CFTR shut. However, it is ineffective against the most common cystic fibrosis mutation. This mutation disrupts CFTR’s folding, preventing the protein from reaching the cell surface. Researchers reasoned that they could solve that problem by combining Kalydeco with a corrector molecule. The corrector would get the channel to its proper place, and Kalydeco would then boost ion flow.
Phase III clinical trial results support this idea. Vertex Pharmaceuticals, maker of Kalydeco, reported on June 24 that a combination of Kalydeco and its corrector molecule, lumacaftor, improved lung function and other symptoms in people with the common CFTR mutation. On the basis of those two trials, Vertex plans to ask U.S. and European regulatory agencies in the fourth quarter to approve the combination.
But the results, while exciting, weren’t as dramatic as they had been for Kalydeco-only treatment of the rare mutation. Biotech analyst Mark Schoenebaum of ISI International puts it in baseball terms: “Kalydeco was a home run. This was a double.”
The reason for these different outcomes has to do with how Kalydeco behaves around “corrected” CFTR, according to independent teams led by Gergely L. Lukacs of Canada’s McGill University and Martina Gentzsch of the University of North Carolina, Chapel Hill. Studying the combination therapy in lung cell cultures, the teams found that in the presence of lumacaftor, or another corrector called VX-661, Kalydeco destabilizes the channel protein, compromising the corrector’s impact (Sci. Transl. Med. 2014, DOI: 10.1126/scitranslmed.3008680 and 10.1126/scitranslmed.3008889).
If these observations in cells translate to animals or humans, patients might benefit even more from different drug combinations, says Raymond A. Frizzell, a cystic fibrosis expert at the University of Pittsburgh.
“While these and other preclinical experiments represent interesting cell biology that can inform our research efforts, what doctors and patients care about—and what we’re focused on—is the benefit the [Kalydeco/lumacaftor] combination has on people with cystic fibrosis,” Vertex spokesman Zachry A. Barber says. A team of Vertex scientists, in collaboration with Frizzell and others, has demonstrated that the combination improved CFTR activity in cells better than either drug alone (Proc. Natl. Acad. Sci. USA 2011, DOI: 10.1073/pnas.1105787108).
Although a rare disease, cystic fibrosis has proven to be a highly lucrative niche for Vertex. Last year, Kalydeco brought in $371 million in sales. Stock analysts forecast peak sales of Kalydeco/lumacaftor to exceed $3 billion.
The power is now in your (nitrile gloved) hands
Sign up for a free account to get more articles. Or choose the ACS option that’s right for you.
Already have an ACS ID? Log in
Join the conversation
Contact the reporter
Submit a Letter to the Editor for publication
Engage with us on Twitter