Advertisement
Grab your lab coat. Let's get started
Welcome!
Welcome!
Create an account below to get 6 C&EN articles per month, receive newsletters and more - all free.
It seems this is your first time logging in online. Please enter the following information to continue.
As an ACS member you automatically get access to this site. All we need is few more details to create your reading experience.
Not you? Sign in with a different account.
Not you? Sign in with a different account.
ERROR 1
ERROR 1
ERROR 2
ERROR 2
ERROR 2
ERROR 2
ERROR 2
Password and Confirm password must match.
If you have an ACS member number, please enter it here so we can link this account to your membership. (optional)
ERROR 2
ACS values your privacy. By submitting your information, you are gaining access to C&EN and subscribing to our weekly newsletter. We use the information you provide to make your reading experience better, and we will never sell your data to third party members.
Analytical Chemistry
Mass spec weighs in on protein therapeutics
Biopharma is applying the high-resolution technique to all points along the drug development pathway
by Celia Henry Arnaud
May 30, 2016
| A version of this story appeared in
Volume 94, Issue 22

Proteins—especially antibodies—are gaining in popularity in the pharmaceutical industry, both as drugs in their own right and as targeting agents for other drugs. In fact, these so-called biologics have been part of the therapeutic landscape for so long that some have already come off patent. Last year, the U.S. Food & Drug Administration approved the first generic, or biosimilar, version of a biologic drug—Zarxio, a version of the bone marrow stimulator filgrastim made by Sandoz. And just last month, FDA approved Hospira’s Inflectra, a biosimilar version of the autoimmune disease treatment infliximab.
In brief
Protein drugs are an ever-growing part of the therapeutic marketplace. But they’re not as easy to analyze as small-molecule drugs. Companies are turning to mass spec to monitor the quality of protein drugs, rather than relying on strict control of the processes used to manufacture them. Read on to learn some of the ways that pharma companies are using mass spec throughout the protein drug discovery and development process.
Protein drugs, though, aren’t as simple to characterize as the small-molecule drugs that sit in most people’s medicine cabinets. Making sure that a batch of protein drugs is highly uniform or that a generic version duplicates an original biologic is a much more complex task than it is for small-molecule therapeutics.
All of that means that pharma companies need high-resolution analytical methods that can catch minor differences in proteins. Of the many analytical techniques available to the industry, mass spectrometry is one of the most versatile. It allows companies to identify proteins on the basis of their amino acid sequences and gives them information about those proteins’ higher-order structures. It allows them to assess a protein’s purity. And in the right circumstances, it can tell them many of these attributes simultaneously.
Mass spectrometry has been wending its way along the protein drug discovery and development path. Initially, it was confined to the early stages of drug discovery, during which scientists use conventional proteomic techniques to identify proteins. But its use has grown. Now it is used in all stages of development to make sure manufacturing processes are pumping out the correct protein and to catch ways in which that protein is being modified during manufacture and storage.
Perhaps the surest sign of mass spec’s growing popularity among pharma firms is that companies are now talking about putting mass spectrometers in pilot plants. “There are groups that are trying to use mass spectrometry to replace some of the traditional quality control assays that you see for biologics,” says Michael T. Boyne II, a mass spec expert and consultant with BioTechLogic. That’s possible, Boyne adds, because mass spectrometry can detect slight changes in protein drugs with high specificity and sensitivity.
As mass spec finally makes inroads into manufacture and quality control of protein drugs, the technique has already cemented its place in the earliest stages of protein drug design. For example, scientists use mass spec to help pick the clone—that is, the DNA sequence—that will produce the protein. That DNA is inserted into mammalian cells, which then make the drug from this blueprint. “We’re essentially screening the top handful of clones” with mass spec to make sure they’re producing proteins with the right amino acid sequence, says Lisa Marzilli, a mass spectrometrist at Pfizer, in Andover, Mass.
Even after the clone is selected, companies must monitor vigilantly for sequence variants. The cells that pharma firms use to produce protein drugs aren’t perfect. Their machinery can naturally create variants by mutating the original DNA sequence, by incorporating the wrong amino acid into the protein drug during translation, or by carrying out other processes, such as posttranslational modification.
When amino acid misincorporations happen, they often indicate a problem with the cell culture medium, Marzilli says. “If instead of asparagine, you have widespread changes to serine, it may mean you ran out of asparagine” in your culture medium, she says. The cell culture engineers can use mass spec data to “reassess their media components and make sure they don’t run out of any particular amino acid. It may be something pretty straightforward and easy to fix.”
“Typically, protein therapeutics have thousands of molecular variants,” says Li Tao, a mass spectrometrist at Bristol-Myers Squibb. Those variants can exist at very low concentrations and have no effect on the drug’s performance. Or they can be abundant and prevent the drug from working properly.
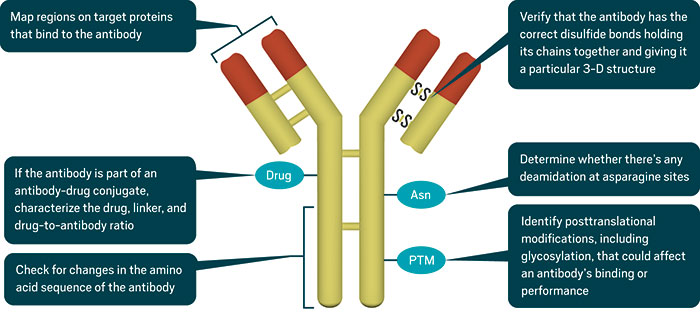

1. Map regions on target proteins that bind to the antibody
2. If the antibody is part of an antibody-drug conjugate, characterize the drug, linker, and drug-to-antibody ratio
3. Check for changes in the amino acid sequence of the antibody
4. Verify that the antibody has the correct disulfide bonds holding its chains together and giving it a particular 3-D structure
5. Determine whether there’s any deamidation at asparagine sites
6. Identify posttranslational modifications, including glycosylation, that could affect an antibody’s binding or performance
Understanding the structure-function relationship of these variants and what processes—either inside or outside the cells—created them “is crucial for the establishment of an appropriate process control strategy and for developing a robust manufacturing process,” Tao says. “Mass spectrometry is deeply involved in the majority of these studies.”
One common variant-generating process is deamidation. In this spontaneous degradation reaction, an asparagine on the protein drug loses an NH3 group and cyclizes to form a succinimide intermediate. Some succinimide is then hydrolyzed to form aspartic acid or isoaspartic acid, depending on where the ring cleaves. Both possible products are 1 dalton heavier than the original sequence. Such a mass shift at the peptide level is easily detected by today’s mass spectrometers.
Depending on the location of a deamidation site within a protein drug, it can actually affect how the protein binds to its target, influencing performance, Marzilli says.
Some amino acid sequences are more prone to deamidation than others. Common “hot spots” include asparagine adjacent to glycine, serine, or threonine. Marzilli and her coworkers are currently evaluating every antibody Pfizer has ever worked with to see if they can assemble a more comprehensive list of troublesome sites to avoid.
But not all deamidation needs to be avoided, Marzilli says. “Say you have 2% deamidation. If it has no effect on the activity of your molecule, it’s just a characteristic of your molecule,” she says. Complete eradication of deamidation is practically impossible, she adds.
Meanwhile, some deamidation sites are harder to find than others. At Genentech, Taylor Zhang found a previously unrecognized trouble spot in the portion of antibodies called the fragment crystallizable region. This region, which is similar across all therapeutic antibodies, is part of the “stem” on the molecules, which have a Y-like shape.
The new hot spot revealed itself when Genentech scientists tested thermally stressed samples. After storing the drug at 40 °C for four weeks, the researchers saw an unusual peak in the drug’s cation exchange chromatogram. “We spent quite some time figuring out what this degradant is,” Zhang says. Mass spec helped uncover clues.
Because this particular deamidation site occurs in a region that’s common across all antibodies, Zhang explains, the knowledge can be applied more generally to antibody development.
In addition to being used to identify sequence and other variants, mass spectrometry is also providing spatial information about therapeutic proteins. For instance, scientists want to know where the binding sites are on their drugs, and they want to make sure the proteins they’re producing have the same structure batch-to-batch and after process changes.
The most common mass spec method being used for such spatial analyses is hydrogen-deuterium exchange (HDX). In HDX, a protein is incubated in a deuterated buffer. During the incubation, deuterium from the buffer replaces amide hydrogens in the protein backbone. After the reaction is quenched, the protein is cleaved into peptides. During mass spec analysis, the extent of deuterium uptake is determined by comparing the masses of these peptides to peptides generated from an identical, nondeuterated protein. Where the deuterium gets incorporated into the protein provides information about how the protein was folded or what it was bound to.
A key application of HDX in the early stages of drug discovery—at least for antibodies—is identifying epitopes. An epitope is the region on an antibody’s target to which the antibody binds.
Epitope mapping “is an important aspect of developing new protein therapeutics,” says Guodong Chen, a mass spectrometrist at Bristol-Myers Squibb. It helps with figuring out the mechanism of action of a particular antibody and helps select the right antibody for a particular target.
To find epitopes using HDX, researchers are looking for regions with significantly reduced deuterium uptake upon binding. That’s because HDX happens to the greatest extent at residues that are solvent accessible and not otherwise engaged in binding. Therefore, amino acids in the epitopes are no longer available to take up deuterium.
Mapping a promising antibody’s epitope can have significant business consequences: If it reveals an epitope that has been claimed by somebody else, the company may have to find another antibody. “Epitope information is crucial for the protection of intellectual property,” Chen says.

There are other methods for epitope mapping, including X-ray diffraction and nuclear magnetic resonance. Both of those methods have their challenges, Chen says. With X-ray diffraction, the antibody and its binding partner, the antigen, can be difficult to crystallize, and NMR is typically restricted to small proteins. Both methods are also slow and require large amounts of material. “Mass-spec-based techniques such as HDX offer unique advantages in sensitivity, specificity, and speed of analysis,” Chen says, although he acknowledges that identifying all the residues involved in epitopes made of amino acids that are not adjacent in a protein’s primary sequence can be challenging.
At later stages in the protein drug development process, HDX-based structural studies can be used for biosimilarity studies. For instance, a company might undertake such a study to show the comparability of a generic protein therapeutic to one that’s already on the market.
“We run HDX as part of our routine testing,” says George Bou-Assaf, a mass spectrometrist at Biogen. “When you are able to provide information at the peptide level that your protein is comparable to a reference standard, it adds a lot of value to your comparability package.” Every time Biogen has an upcoming filing with FDA, whether it’s for an amendment to an Investigational New Drug Application or for a process change, the company runs these comparability studies, Bou-Assaf explains.
All the mass spec analyses mentioned so far are for a specific antibody or protein therapeutic on its own. But antibodies are increasingly being used as targeting agents for other drugs. These so-called antibody-drug conjugates, or ADCs, have to be characterized too—both the combination and each piece on its own.
“It’s four times the amount of work,” Pfizer’s Marzilli says. “You need to characterize the payload, which is the drug. You need to characterize the linker, which attaches the payload to the antibody. You need to characterize the antibody. And you need to characterize the antibody-drug conjugate.”
“The structural complexity of ADCs has required us to come up with completely new analytical strategies for trying to understand the safety and efficacy of these molecules in preclinical studies and clinical trials,” says Surinder Kaur, an associate director in the bioanalytical sciences department at Genentech. For normal therapeutics, she says, the pharmacokinetics usually reflects decreasing concentrations of the drug as it is metabolized over time. “With ADCs, because there are multiple species, there’s a lot of strategy involved in what species to measure, and we combine immunoassay and mass spec technologies to measure the concentrations.”
For such analyses, the antibody and the conjugate are usually run side by side so the scientists know what the antibody looks like on its own. At Pfizer, Marzilli is double-checking the antibody’s amino acid sequence, or primary structure, and attachment sites. But mostly, “we’re looking for anything we’re not expecting,” Marzilli says. “We’re looking for consistency from material to material. We’re looking for everything-plus with an ADC.”
So far, mass spec in biopharma has been confined to research and development. But there are efforts afoot to push it into the realms of manufacturing and quality control.
Richard Rogers, a scientist at Just Biotherapeutics, is leading a consortium of biopharma and instrument companies with the goal of putting a mass spectrometer on the pilot-plant floor. “We envision the mass spec being an essential component for process development,” Rogers says. “We’re already using it in the small-scale labs. But if we can use it in the pilot plant, we will know what our product looks like before it even makes it to downstream purification.”
What could help that happen is a mass spec method that allows scientists to monitor multiple attributes of protein drugs—amino acid sequence, structural integrity, and the like—simultaneously and detect new modifications or impurities that show up as new peaks (mAbs 2015, DOI: 10.1080/19420862.2015.1069454). Rogers helped develop the method when he was a scientist at Amgen. Classical analytical assays—such as cation exchange chromatography (CEX) and capillary electrophoresis (CE)—typically tackle one attribute at a time. But process monitoring requires looking for many attributes simultaneously. “Any sort of posttranslational modification you can imagine creating a mass shift or a chromatographic retention time shift of that molecule can be monitored using mass spectrometry,” Rogers says.
Advertisement
The goal is to use mass spec to monitor the quality of protein drugs instead of relying just on strict control of the processes used to manufacture them. “We are defining what we want our product to look like and then we are affecting the cell culture and the downstream unit operations to give us that molecule,” Rogers says.
The multiattribute method is intended to replace less specific methods such as CEX and CE. “We plan on using this method during development because it is more specific in terms of the chemical modifications and their positions in the molecule,” says Rohini Deshpande, executive director of process development at Amgen. “The method allows a great number of modifications to be monitored during development, before the biological understanding of the attributes is known.”
The most important part of the method is its ability to catch impurities in the protein by detecting unexpected peaks in the mass spectrum. As Rogers runs his tests, he’s noted that the only instrument able to consistently detect these new peaks is the Orbitrap, a high-resolving-power mass analyzer made by Thermo Fisher Scientific, he says.
And that’s the reason for including instrument companies in the consortium. Rogers hopes that sitting in on the discussions of what biopharma companies need will “spur the vendors on to create better software and better mass specs to do this more efficiently.” He would love to see a mass spec with the resolving power of an Orbitrap in a box small enough to sit on top of a chromatography tower.
Amgen is the furthest along in adopting the multiattribute method. The company has “proactively engaged” FDA in discussions about implementing the method. “The Amgen strategy includes a phased implementation starting with the inclusion of the multiattribute method as a characterization assay and then as a release assay,” which is a test run to make sure a drug batch meets product specifications, Deshpande says.
Mass spec is an evolving area in quality control, says BioTechLogic’s Boyne. “There’s an interest in moving mass spectrometry into QC to simplify and minimize the number of release assays that are run,” Boyne says. “It’s a goal. We are making progress within the community to make it a reality.”

The power is now in your (nitrile gloved) hands
Sign up for a free account to get more articles. Or choose the ACS option that’s right for you.
Already have an ACS ID? Log in
Join the conversation
Contact the reporter
Submit a Letter to the Editor for publication
Engage with us on Twitter