Advertisement
Grab your lab coat. Let's get started
Welcome!
Welcome!
Create an account below to get 6 C&EN articles per month, receive newsletters and more - all free.
It seems this is your first time logging in online. Please enter the following information to continue.
As an ACS member you automatically get access to this site. All we need is few more details to create your reading experience.
Not you? Sign in with a different account.
Not you? Sign in with a different account.
ERROR 1
ERROR 1
ERROR 2
ERROR 2
ERROR 2
ERROR 2
ERROR 2
Password and Confirm password must match.
If you have an ACS member number, please enter it here so we can link this account to your membership. (optional)
ERROR 2
ACS values your privacy. By submitting your information, you are gaining access to C&EN and subscribing to our weekly newsletter. We use the information you provide to make your reading experience better, and we will never sell your data to third party members.
Computational Chemistry
C&EN 中文版
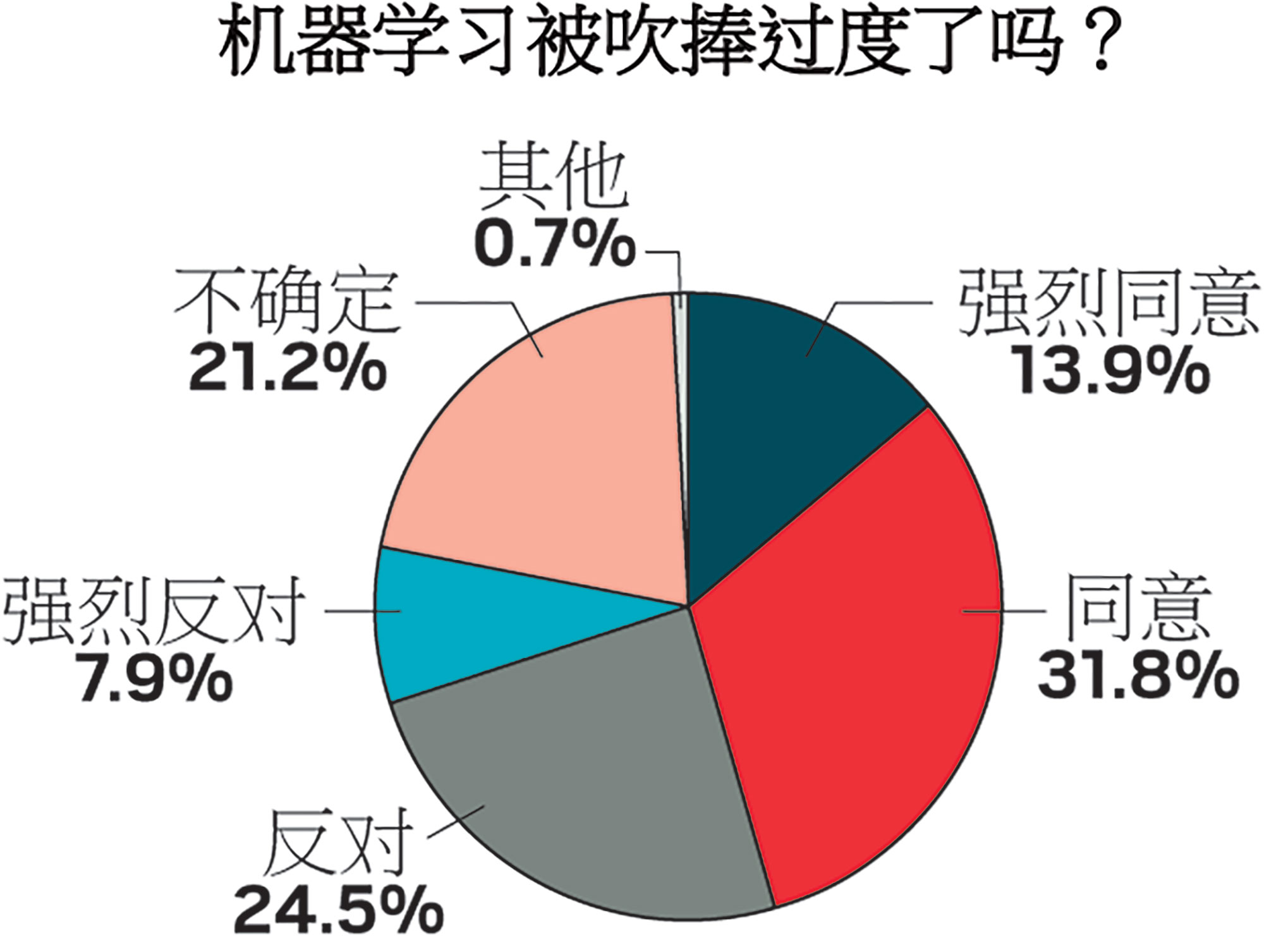
机器学习被吹捧过度了吗?
化学家们点评该技术的潜力和陷阱
by Sam Lemonick
August 27, 2018
| A version of this story appeared in
Volume 96, Issue 34

Credit: Shutterstock/C&EN
机器学习完全被过度吹捧。机器学习完全没有被过度吹捧。机器学习是变革性的。机器学习同其他技术一样仅仅是工具而已。
如果询问10名化学家如何看待机器学习的潜力,你将会得到十个不同的答案。这可在推特上引起生动的辩论,但讨论本身具有严肃的意义。
来自美国西北大学的物理化学家George Schatz认为,如果机器学习没有所宣称的那么有价值,那么在实验室进行试验的“人们在浪费时间和精力”。如果该工具无法如承诺般解决问题,那些在机器学习上投入培训、实验时间和经费的科学家们可能会发现自己身处困境。
另一方面,如果机器学习是未来的潮流,那么尚没有使用机器学习的化学家们将面临落后于同行的风险。
对于“机器学习是否被过度吹捧”这一问题,也许目前不可能有一个肯定的答案。但是,C&EN在和化学家们进行了数十次对话之后,得出了有关于当前机器学习现状的共识。
机器学习是人工智能的一种,它是计算机使用一组数据进行训练后基于该数据建立规则或者认知的功能。化学家们往往对于该工具的预测能力感兴趣。比如,当我们在一个机器学习算法中输入100种合金及其熔点信息的列表时,它能否能对未出现合金的熔点进行预测?甚至可能对一种从未合成过的合金的熔点进行预测?
尽管机器学习有很多潜力——或貌似潜力,但有一件事情是确定的,那就是机器学习不是神奇的。“让我们现实些,”来自谷歌的计算机科学家 George Dahl说,“机器学习是非线性回归,”是一种使用收集数据生成模型参数的简单统计分析。Dahl在多伦多大学的Geoffrey Hinton实验组的研究生学习期间曾在默克公司机器学习竞赛中获胜。
把机器学习弄的名不符实,会对技术本身不利。如果它无法达到预期,资助者和科学家们可能会得出机器学习不值得浪费时间的结论。“我们需要吸引最聪明的人才”去研究它(机器学习)、探索其裨益,从而让其成功,维也纳大学的有机合成化学家Nuno Maulide表示。
把机器学习弄的名不符实,会对技术本身不利。如果它无法达到预期,资助者和科学家们可能会得出机器学习不值得浪费时间的结论。“我们需要吸引最聪明的人才”去研究它(机器学习)、探索其裨益,从而让其成功,维也纳大学的有机合成化学家Nuno Maulide表示。
药物发现
基于多种原因,制药公司自然称为了机器学习的早期使用者。当许多化学家尚且无法接触到机器学习的时候,医药公司却可以提供和承担计算能力,而且他们拥有大量有关小分子和生物靶点的数据可用于算法训练。
该产业除了资源外还有动力。与组合化学和高通量筛选(HTS)一样,机器学习给出了可以加速和改善药物发现的承诺,而组合化学和HTS的光辉已经随时间黯淡。“因为药物的开发极为困难,故制药公司对于新技术满怀希望,且从业者往往寻求新的途径获得成功。”计算化学家Robert P. Sheridan说,他近期刚从美国制药公司默克退休。
调查结果
C&EN对化学家们关于机器学习的观点进行了在线调查。以下为调查结果。
30%
的受访者说他们经常在工作中使用机器学习。
什么是机器学习有潜力产生最大影响的化学领域?
“就我个人经验而言,由于信息的海量存在,我们很难确保从文献检索中获取的结论毫无偏颇。我认为机器学习将帮助我们获取统计上无偏见的文献信息。” ——伯明翰大学研究生Samuel Nunez-Pertinez
它有潜力显示出那些人类倾向于忽略的数据趋势。机器学习所提供的那些工具可提供一些能激发新想法的建议,从而帮助化学家们作出决策。——麻省理工学院博士后 Thomas Struble
哪些化学领域对机器学习抱以的期待是最不现实的?
“因为机器学习仅仅是大数据集中的插值,在那些难以生成大量可靠数据的领域将依然很难或不能使用它,因为机器学习的外推法将会制造极为错误的答案。” ——基尔大学教授Bernd Hartke
“分析化学。对于化学家们和立法者而言,在不知道从样本到结果的确切过程的前提下,仅依赖机器学习的结果是很难工作的。” ——新加坡国立大学博士后Andrea Leoncini
“正如密度泛函理论(density functional theory)没有取代波函数方法(wave-function-based methods),也没有消除进行实验的需要,机器学习也将无法取代量子化学方法。但是它确实有潜力带给我们的运算更好的回报。” ——佐治亚大学研究生Marissa Estep
你所从事的化学领域?
有机化学:
23.3%物理化学:
17.3%
分析化学:
16.0%无机化学:
12.7%
生物化学:
10.0%计算化学
7.3%
其他
13.3%总人数= 150人
备注:因四舍五入,总和不等于100%。并非所有受访者都对每个问题作出了回答。
来源:2018年C&EN在线投票
制药公司第一次使用机器学习的确切时间,取决于我们对机器学习的定义。在药物设计中使用了人工神经网络的算法已经近半个世纪,这是一种机器学习的简单形式。1973年,前苏联的一个科学团队证明,人工神经网络可以预测1,3-取代的二恶烷(substituted 1,3-dioxanes )的生物活性。(论文来源: Comput. Biomed. Res.1973, DOI: 10.1016/0010-4809(73)90074-8)
自上个世纪九十年代起,药物化学家们在定量-结构活性相关(quantitative structure-activity relationship ,QSAR)模型中使用了人工神经网络算法。定量-结构活性相关模型可以基于已知分子特性对相关分子特性做出预测,从而帮助科学家决定是否将该分子作为可行药物进行开发。同样作为机器学习的类型,随机森林(Random-forest ,RF)算法和支持向量机(support vector machines,SVM)目前很大程度上取代了人工神经网络算法在定量-结构活性建模中的使用。
从这一角度而言, Sheridan认为称机器学习过度炒作并不恰当。“在化学领域,定量-结构活性相关模型意义上的机器学习已经使用了数十年,而且是证明有用的”,他说。定量-结构活性相关模型不是完美的,但是默克等公司仍然使用,因为该模型可帮助化学家们选择哪些分子作为优先考虑,从而节约时间和成本。
医药界已经经历了机器学习的“炒作周期”。美国Gartner咨询公司在1995年描述人们对于新技术的看待方式时,首次使用了该术语。首先,创新会快速引起关注达到过度期望的高峰阶段,之后会沉入泡沫化的谷底阶段。随着人们加深理解其局限性和实际能力,该技术达到生产力的高峰阶段。熟悉制药业的人也许会看出和一些曾被过度吹捧的创新技术——诸如纳米技术和组合化学——类似的阶段演变。
医药公司们看似并未对这些重复出现的炒作产生任何抵抗力。在Sheridan看来,目前药物开发领域的“深度神经网络”正在接近炒作的高峰。同上述的人工神经网络一样,它模拟大脑:将信息通过一系列互相连接的点送至神经元。每个点以特定的方式对数据信息进行分析,然后传递给其临近的点。这是图像识别软件的运行方式,首先识别阴影和形状,然后是眼睛和耳朵,最终实现人脸识别。深度神经网络比之前的类似技术具备更多节点层面,在图像识别和自然语言处理领域产生了巨大的进步。
深度神经网络能从非常复杂的数据中学习,这种能力使其在药物化学领域尤具魅力。美国软件公司Atomwise制造了基于深度神经网络的软件,该软件用于预测候选药物和体内靶标的结合亲和力。公司联合创始人兼首席执行官Abraham Heifets将深度神经网络的引入称为是机器学习领域的一项根本性变革。
有人指责Atomwise公司对机器学习的功能过分夸大。2015年一篇刊发在美国科技媒体TechCrunch的文章引述该公司联合创始人Alexander Levy 的言论,称Atomwise的软件让他在自家客厅便可对麻疹的治疗方法作出预测。药物发现和制药博客In the Pipeline 中针对该文章发表了一条帖子,Levy对该帖回复中表示部分责任归咎于该文的记者:“假如Atomwise有TechCrunch文章所述的一半那么棒,该有多好?”他于今年早些时候离开了Atomwise公司。
Abraham Heifets看起来认识到Atomwise这类公司的口碑。他澄清道,Atomwise并不对任何治疗进行预测。“我们的预测的是结合亲和力,结合亲和力不是药物,”他说。
该领域的其他人也响应了这一观点。即使机器学习可以准确预测出与靶点高度结合亲和的分子,即使机器学习可以做的比人类更好,但是这离创造出可盈利新药尚有很大距离。“新药发现的路径长,”来自谷歌的Dahl说。在这些研究过程中加入机器学习的工具就好比开发出了更好的显微镜或是检测方法,他补充道,这将产生小但实际的影响。
大型制药公司对深度神经网络仍然充满热情,不过和Heifets一样,他们也很小心地调整期望值。“设想人工智能或是机器学习可以解决所有的问题,这并不是我们思考这些技术的方式。它们在一些单独任务的完成上是强有力的,”Jeremy Jenkins说,他是诺华公司化学生物和疗法数据科学的负责人。
尽管如此,化学家们将深度神经网络视作把药物发现提高至新水平的一条途径,因为它可以处理和诠释人体内部的复杂生物数据。“通常,生物学是如此复杂、让人很难理解,” Vijay Pande说,他是美国风险投资公司 Andreessen Horowitz的合伙人,斯坦福大学的计算化学家。他认为人类对于生物数据的理解已经到达极限,而机器学习可以弄懂人体生物学和医药化学之间的介面。
Jeremy Jenkins表示这一功能使机器学习可以向医药化学家们建议一个或数个分子,让他们更好地集中力量,从而更快地开发出潜在药物。这听起来很像早期机器学习的倡导者所作出的许诺,而有人怀疑深度学习也是一样。Sheridan 说,当他的研究组将深度神经网络与其他机器学习方式对比,他们发现该方式的预测能力在统计学上有显著提升。但是他也表示,在整个药物发现流程中,收益是是有限的。
Advertisement
也许,虽然机器学习能够造福新药开发,但将会对其他化学领域产生更大的影响。
材料研究
“在药物发现领域使用机器学习很难,其原因在于问题本身不明确”, Leroy Cronin说,他是英国格拉斯大学的一名化学家。在具备明确的目标前提条件下,机器学习已经证明可以做得很好,例如脸部识别。Cronin 解释道,因为我们并不完全了解药物成功的因素,我们不知道在使用机器学习时,使用哪些数据可以使其获得成功。对比之下,我们完全了解构成人的面部的元素。也许深度神经网络处理复杂数据的能力可以将这些数据区分开来,但是仍然有待观察。Cronin等人认为机器学习在材料研究中可能会更快地产生更大的影响。
“化学如何影响物质的属性?其中有着直接的关联,”Jillian Buriak说。比如,晶体的硬度取决于原子间互相结合的方式。她解释说,这意味着机器学习或许对材料研究更有用。Jillian Buriak是加拿大阿尔伯塔大学的材料化学家,也是《材料化学》(Chemistry of Materials)期刊的主编。
材料科学仅在过去十年间使用机器学习,晚于医药化学领域。但是,已有一些论文证明机器学习算法可对分子或具备所需特性的材料进行预测,一些结果令人惊讶。比如,机器学习算法发现了自旋交叉配合物(spin-crossover complex),这是一种可用来作为开关和传感器的无机复合物(论文来源:J. Phys. Chem. Lett. 2018, DOI: 10.1021/acs.jpclett.8b00170);他们还发现了使用合金制造的金属玻璃,在此之前并未形成理论(论文来源:Sci. Adv.2018,DOI: 10.1126/sciadv.aaq1566)。
和制药业一样,材料研究领域长期以来使用了一些技术,这些技术在今天被称为“机器学习”。麻省理工学院的化学工程系教授Heather Kulik主导了上述自旋交叉配合物的研究,忆及四、五年前开始组建自己的实验室时,她告诉大家自己的目标是:将有机化学信息学的功用拓展至无机化学。“现在我永远不会那样说了”,Kulik说,“现在我会说,我们借助机器学习加速对无机化学的探索,因为这更能获得研究资助者和论文读者的响应。”
Buriak 同样认为材料研究者们已经默默地使用机器学习多年,虽然他们并未以此称呼它。他指出,类似材料基因组计划(Materials Genome Initiative,MGI)的一些项目便是如此。该项目是资金超过5亿美元的多个联邦机构合作的项目,于2011年启动,目标是更快地发现和制造新材料。该项目收集并分享了材料数据从而加速材料科学的进步,尤其是借助了计算机。
材料基因组的概念可以追溯至2002年,当时的设想是大量收集和分析数据以产生新的启发,2016年科研人员们在一个展示机器学习可以裨益材料科学的示范试验中使用了它。研究者们建立了一个包含有失败试验数据的公共数据库,可以比人类更准确地预测钒亚硒酸盐结晶反应。(论文来源: Nature 2016, DOI: 10.1038/nature17439)
计算机可以在一些任务中击败人类是一回事儿,不过使用机器学习的研究者们有一个共识——计算机无法取代人类的直觉。美国橡树岭国家实验室的物理化学家 Bobby G. Sumpter从20世纪八十年代以来一直从事算法研究,他表示,有理由相信机器学习可以插值大量数据和作出预测,这些预测对于人类科学家们而言太过于微妙和复杂。但如果认为机器学习可以对远远超出特定训练数据的化学领域作出预测,那就是无视统计的基本原理,Sumpter说。
如果这是可行的,那么举例而言,我们可以从一个分子的可观测属性对其结构作出推测。但目前这并不可能实现。
Bert de Jong是来自美国劳伦斯伯克利国家实验室的一名计算化学家。在他看来,我们现在所说的机器学习是用于对大数据集中运算加速的工具。它不能够超出数据集提供的信息进行推算、了解分子的物理特性、并进行真正的学习。
相反,许多人认为机器学习可以通过改进实验对材料化学家提供帮助。Sumpter 说,“机器学习能大力帮助分析和提供有意义的结果。”
在他看来,在图像解释、化合物材料的光谱,尤其是在仪器检测限值的噪音中寻找信号这些方面,机器学习已经彰显了其优势。他表示,机器学习还可在实验中进行实时引导。因为机器学习仅需数毫秒便可以吸收和解读庞大的数据,人工智能可以对输入和参数进行调整,从而在实验过程中提供实时优化,尤其是流式反应装置中。
如果能够实现良好构建,机器学习不会受人类的偏见的影响,也更前后一致。对许多人而言,这也使机器学习更适合设计实验。“化学是杂乱和复杂的”,Cronin 说,“机器学习可以帮助设计更好的实验,”尤其是当变量的数量超出人类所能,如了解溶剂对于反应的影响。
反映发现
当许多化学家听到“机器学习”和“炒作”这些词汇时,第一个闪现在脑海的是“逆合成”。美国哈佛大学的有机化学家Elias J. Corey发展了逆合成分析理论,几乎从他第一次描述借助关键化学键和合成砌块制定目标分子的合成路径的概念以来,他和其他研究者便一直努力打造可以设计合成路径的计算机程序。
“他们坚持说了那么久,所以每个人的对此都是确信不疑,”Sumpter 说。尽管过去曾持有怀疑, Sumpter也认为机器学习可以通过计算机实现合成。而身处该领域的化学家们谈论机器学习的方式比大多数人预期的更为低调。
“我视机器学习为提升人类能力,不是让人类变得不必要,而是让人类在所做的所有事情中更为有效率,” Matt Toussant说,他是美国化学会(American Chemical Society)旗下分支机构美国化学文摘社(Chemical Abstracts Service,简称“CAS”)的产品与内容运营高级副总裁。美国化学会出版 C&EN。
据Toussant介绍,美国化学文摘社在今年秋天将推出名为“ChemPlanner”的逆合成规划产品。八月份,德国制药巨头默克公司的子公司 MilliporeSigma发布了名为“Synthia”(此前名为 Chematica )的软件,这是该领域另一个重要的产品。
上述两个计算机程序都强烈依赖着人类专家建立的数据库,他们创建了化学转化必须遵守规则的数据库,这些来自于文献和他们自己的知识。机器学习算法使程序利用这些规则探索化学空间,并向使用者建议合成目标分子的可行方法。
韩国蔚山国立科学技术研究所的Bartosz Grzybowski 教授是“Synthia”的创造者,他认为机器学习仅仅是软件所依赖的工具之一。“Synthia”还使用分子动力学、量子力学和电子性质来判断转化的好处或合成路径上中间体的稳定性。机器学习不是万能的, Grzybowski 教授说,“有机化学中一些超前部分需要这些工具。所以我倡导开放,解决问题,而不要固守单一模式。”
一些科学家仍怀疑这些产品是否将超越传统的合成路径规划的方式,提供显著的优势。规划合成路径的传统方式是,研究生使用诸如“Reaxys ”或 “SciFinder”(两者均为美国化学文摘社旗下产品)等工具访问数据库,推理出一条规划路径,并进行试验。Toussant 表示,化学家们关心的是“ChemPlanner”这样的机器学习算法能否使他们更高效且更多发现。
Grzybowski用自己的口头禅回应这些疑虑:“去做!” ,即用“Synthia”所给出的合成路径在实验室进行试验,将其所得路径与人类使用数据库得出的路径相比,是否更优或在更短时间内获得同样路径。他在2018年发表了试验结果,(论文参见: Chem2018,DOI: 10.1016/j.chempr.2018.02.002),结果显示“Synthia”可在15或20分钟内给出新的、有效、可行的合成路径。

图片:基于机器学习的“Synthia”规划出一条用于ATR 抑制剂的合成路径(右图),该路径与一条发表的路径(左图)相比减少了几个步骤但收益相似。
如果化学家们认为需要使用“机器学习”这类时髦用语吸引眼球或是金钱,Grzybowski并不责怪他们。“影响因子是上帝”,他说。但他表示,一旦机器学习的炒作结束,有价值的工具将会留下,如此前大热的组合化学或基因组学,两者均证明了这一点。它们都上过炒作的“过山车”,虽然最终并未兑现一些人的许诺,但是都仍在使用中。
Toussant认为现在已接近机器学习炒作的顶峰,将要滑落幻灭的谷底。“不过最终所有技术都会从绝望的深渊中翻身,”他说,“我期待机器学习也是如此。我相信它的未来。”
人类学习
尽管观点有所差异,C&EN所采访的科学家们均认同:是的,机器学习是被过分炒作了。不,它不能治愈癌症。但尽管如此,它是一个值得保留的有价值的工具。
其价值究竟多大,每个人的看法不同。但为了让化学家们充分利用机器学习,有一件事情是肯定的。即,化学家们需要转变他们的行为习惯,从数据开始。
医药和材料科学这两个化学领域已致力创建化学特性的有用数据库,在机器学习领域走得最远,这并不奇怪。获得纯净、全面的数据以建立训练样本库,这是将机器学习应用于有机合成领域的一个难题。成功试验的数据以各种形式和注记散布在众多期刊里,而那些失败试验的信息则藏在实验室的旧记录本上。
“旧的准则仍然适用,”美国橡树岭国家实验室的 Sumpter说,“废料入,废料出(质量差的输入产生质量差的输出)”。当对大量有组织的数据进行训练的时候,最好也包括负面结果的数据,在这种情况下机器学习的效果最好。格拉斯哥大学的 Cronin认为,化学家们需要学习如何建立数据库和创建数据的描述符,以便于算法学习。
化学家们还将需要基本的编程技能。西班牙阿利坎特大学的无机化学家 Javier García Martínez认为,需要改变化学家的培训。“每一个博士生都知道核磁共振和X射线衍射”,他说,“新的工具将是机器学习和人工智能。”对于已经完成正式训练的化学研究者们,García Martínez 鼓励他们使用免费在线工具进行自学。
许多人认为,合作也将会变得越来越重要。有机化学家将会与计算化学家或计算科学家们合作,寻找将机器学习应用到他们研究中的方式。Dahl 表示希望有更多的化学家带着他们的数据和问题前往谷歌,“我很高兴尝试进行合作。”
如果不真正地努力让分子科学家们学习新技能、改变他们对数据的思考方式、甚至以不同的方式提问,即使是最坚定的机器学习支持者也不会相信机器学习对化学家会有用。美国福坦莫大学和哈弗福德学院的计算化学家Joshua Schrier认为,如果科学家们可以做到上述几点,“机器学习可以赋予普通化学家超级能力。”
机器学习是否被过度追捧取决于看待它的角度。对那些与机器学习密切相关的化学家们而言,新闻稿或日常谈话中对此话题的兴奋感可能会变得令人厌倦。这些专家们对机器学习能够真实且有效影响化学具有高度的信念,尤其是如果有更多人接受训练有效使用。与此同时,一些人担心这一工具并不能兑现高度期望,而失望则会阻碍前进。
Cronin 这样表述:“虽然我认为机器学习被过度追捧,这点很恼人,但我认为化学家们还没有充分利用它。”
Chemical & Engineering News
ISSN 0009-2347
Copyright © 2024 American Chemical Society
You might also like...



The power is now in your (nitrile gloved) hands
Sign up for a free account to get more articles. Or choose the ACS option that’s right for you.
Already have an ACS ID? Log in
Join the conversation
Contact the reporter
Submit a Letter to the Editor for publication
Engage with us on Twitter