Advertisement
Grab your lab coat. Let's get started
Welcome!
Welcome!
Create an account below to get 6 C&EN articles per month, receive newsletters and more - all free.
It seems this is your first time logging in online. Please enter the following information to continue.
As an ACS member you automatically get access to this site. All we need is few more details to create your reading experience.
Not you? Sign in with a different account.
Not you? Sign in with a different account.
ERROR 1
ERROR 1
ERROR 2
ERROR 2
ERROR 2
ERROR 2
ERROR 2
Password and Confirm password must match.
If you have an ACS member number, please enter it here so we can link this account to your membership. (optional)
ERROR 2
ACS values your privacy. By submitting your information, you are gaining access to C&EN and subscribing to our weekly newsletter. We use the information you provide to make your reading experience better, and we will never sell your data to third party members.
ACS Meeting News
Structures of new cancer drug candidates revealed
Six companies showed off their oncology efforts in first-time disclosures session
by Brianna Barbu
March 23, 2024
On Wednesday, the popular “First-time disclosures” session of the ACS Spring 2024 meeting, in the Division of Medicinal Chemistry, offered a look behind the curtain of the drug development pipeline with the unveiling of the structures of new clinical candidates. This year, it was cancer across the board as six companies—Oric Pharmaceuticals, Nurix Therapeutics, Schrödinger, Deciphera Pharmaceuticals, Tango Therapeutics, and Blueprint Medicines—presented structures and early clinical trial results for new oncology drug candidates in a hybrid session.
But while all six candidates were connected in terms of the disease they’re targeting, each one tells a different story about problem-solving in medicinal chemistry, said Nicole Goodwin of GSK, who organized and served as moderator for the first half of the session. Rachel Lagiakos of Schrödinger moderated the final three presentations; she’ll be taking over the organizer role from Goodwin in future meetings.
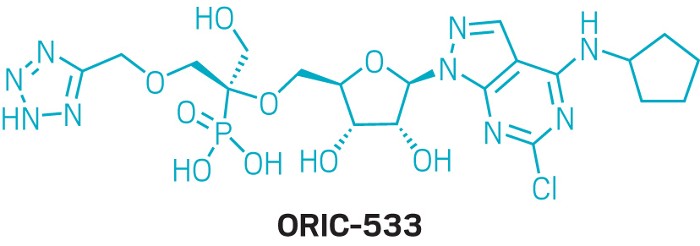
Candidate: ORIC-533
Presenter: Jared Moore, Oric Pharmaceuticals
Target: CD73
Disease: Multiple myeloma

In the first presentation of the session, Moore unveiled ORIC-533, an oral drug candidate designed to inhibit the adenosine-producing enzyme CD73. This enzyme is part of an immune suppression pathway that has been linked to drug resistance in multiple myeloma and other cancers. The researchers’ challenge was to design an inhibitor that would maintain its potency even with high levels of adenosine monophosphate, CD73’s usual substrate, competing to bind to the enzyme.
The core of the molecule is an adenosine derivative; Moore and coworkers focused on maximizing hydrogen bonding while balancing the overall charge and polarity to keep bioavailability high. They found that the area around the phosphonate group, which forms a salt bridge between the protein and a zinc ion, had the biggest impact on how well candidates blocked adenosine production in biochemical assays. The ether-linked tetrazole forms hydrogen bonds with three key amino acids in CD73’s active site to boost the drug candidate’s potency.
ORIC-533 is currently being tested in Phase 1b clinical trials for people with multiple myeloma. It’s one of three small-molecule CD73 inhibitors in active clinical trials. The company plans to partner with Pfizer on a Phase 2 study.
Candidate: NX-1607
Presenter: Frederick Cohen, Nurix Therapeutics
Target: Casitas B-lineage lymphoma proto-oncogene B (CBL-B)
Disease: Cancer

The cancer immunotherapy talks continued with Frederick Cohen presenting NX-1607. This drug candidate was made to target CBL-B, an immune checkpoint protein that limits the ability of T cells to mobilize against tumors. CBL-B is a challenging target because it doesn’t have a well-defined small molecule docking site.
Cohen and coworkers’ starting point was a molecule that had been shown to act as an intermolecular glue, keeping CBL-B folded up in its inactive state. Through high-throughput screening, the researchers found that including N-methyl triazole in the candidate structure provided what they dubbed a “pocket-filling widget.” The triazole’s nitrogens form hydrogen bonds to keep the molecule attached to the protein for longer. A cyclobutane spacer helps the triazole enter the pocket at just the right angle. Meanwhile, a benzylic amine fits into another key hydrophobic pocket on the enzyme.
In preclinical animal studies, NX-1607 worked well to increase T cell activity and fight tumors by itself and in combination with anti-PD1 immune checkpoint inhibitors. It’s highly soluble and permeable, which should make it easy to formulate, Cohen said. Phase 1 clinical trials are ongoing for NX-1607 by itself and in combination with paclitaxel for people with advanced solid tumors.
Candidate: SGR-1505
Presenter: Zhe Nie, Schrödinger
Target: Mucosa-associated lymphoid tissue lymphoma translocation protein 1 (MALT1)
Disease: B-cell lymphoma

Zhe Nie talked about how Schrödinger used their computational screening platform to design SGR-1505, a new potential treatment for B-cell lymphoma. The molecule is designed to deactivate a protein in cancer cells called MALT1 by binding to an allosteric site. MALT1 is an encouraging target for cancers that are resistant to Bruton’s tyrosine kinase (BTK) inhibitors because it’s part of the same signaling pathway as BTK.
Schrödinger uses computational modeling to predict binding affinity between proteins and small molecules, which lets them screen thousands of potential drug structures and select the best ones to make and test in the lab. The discovery effort took about 10 months and involved synthesizing only 129 of the thousands of structures modeled for binding affinity—after more than 8 billion compounds that had been computationally assessed in some way.
SGR-1505 is currently being tested in Phase 1 clinical trials for people with advanced B-cell lymphoma. Schrödinger scientists expect initial data to come out in late 2024 or 2025.
Candidate: DCC-3116
Presenter: Kristen Stoltz, Deciphera Pharmaceuticals
Target: ULK1/2
Disease: Cancers associated with RAS/RAF mutations

Mutations in the receptor tyrosine kinase (RTK) pathway are common in many cancers, so there are many cancer therapies that target RTK proteins. However, cancer cells can resist these drugs using a mechanism called autophagy, which helps them stay alive by recycling damaged proteins. The proteins ULK1 and 2 are responsible for activating autophagy, so the team at Deciphera wanted to come up with a drug to inhibit them.
The molecule they designed, DCC-3116, boasts a central pyrimidine motif with two secondary amines that form a two-armed structure that binds to the protein and locks it in an inactive conformation. There are other drug candidates with similar structures involving a central pyrimidine bound to two amine groups, but DCC-3116 is more potent and slower to dissociate. Stoltz attributed this to optimized interactions between the arms of the molecule and the protein: the methylpiperazine interacts with a key aspartate residue in the protein, and the lactam ring fits just so into a pocket formed by the enzyme’s P-loop.
Based on its effectiveness inhibiting autophagy and fighting tumors in animal testing, DCC-3116 is currently being tested in Phase 1 trials by itself and in combination with RTK inhibitors. Stoltz says that her team hopes to identify a dose to use in Phase 2 trials later this year.
Candidate: TNG348
Presenter: Scott Throner, Tango Therapeutics
Target: ubiquitin-specific protease 1
Disease: BRCA1/2-mutant cancer and other homologous recombination deficient (HRD+) cancers

Scott Throner of Tango Therapeutics presented TNG348, a reversible allosteric inhibitor of USP-1 and a potential treatment for HRD+ cancers. HRD+ cancers make up a large fraction of ovarian and breast cancers as well as some prostate and pancreatic cancers. They also have damaged DNA-repair processes. The Tango team determined that taking out USP-1, another DNA-repair enzyme, would be an effective way to kill these cancers.
Based on previous reports of USP-1 inhibitors in the literature, Throner and coworkers identified guanidine derivatives as a promising path to pursue. They screened a number of benzylic linkers and functional groups on the aromatic group. They also added fluorinated groups to make the core of the molecule less basic and avoid unwanted interactions with cardiac potassium channels. When the molecule binds to an allosteric site on USP-1, it nudges the enzyme’s H-bonding network out of alignment to impede its activity.
Preclinical results showed that TNG348 was effective at shrinking tumors by itself and with PARP inhibitors. It recently entered Phase 1 clinical trials in people with HRD+ breast and ovarian cancer. The first patient received the treatment at the end of last year.

Candidate: BLU-222
Presenter: Philip Ramsden, Blueprint Medicines
Target: Cyclin-dependent kinase 2 (CDK2)
Disease: HR+/HER2- breast cancers and other CCNE1-aberrant cancers
Cyclin dependent kinases (CDKs) help regulate the cell division cycle. CDK2 is a promising drug target for cancers where the gene that encodes for its partner, Cyclin E1, is overactive. That includes HR+/HER2- breast cancers that are resistant to CDK4/6 inhibitors, the current standard of care for those cancers. The Blueprint team focused on making an inhibitor that would be specific to CDK2—a challenge, because its active site is nearly identical to that of CDK1.
Ramsden and coworkers screened Blueprint’s compound library, which is “annotated” with data about each compound’s interaction with the full slate of human kinases, to find a CDK2-selective lead. They found that a pyrazine core gave the best combination of selectivity and potency. A pyrazole ring with a difluoromethoxy group provided a key interaction with the π electrons in a “gatekeeper” phenylalanine residue to further boost selectivity against other kinases. The result was a molecule, BLU-222, that’s 90 times more selective for CDK2 than CDK1. BLU-222 is now undergoing Phase 1 clinical trials by itself and in combination with CDK4/6 inhibitors.
You might also like...




The power is now in your (nitrile gloved) hands
Sign up for a free account to get more articles. Or choose the ACS option that’s right for you.
Already have an ACS ID? Log in
Join the conversation
Contact the reporter
Submit a Letter to the Editor for publication
Engage with us on Twitter