Advertisement
Grab your lab coat. Let's get started
Welcome!
Welcome!
Create an account below to get 6 C&EN articles per month, receive newsletters and more - all free.
It seems this is your first time logging in online. Please enter the following information to continue.
As an ACS member you automatically get access to this site. All we need is few more details to create your reading experience.
Not you? Sign in with a different account.
Not you? Sign in with a different account.
ERROR 1
ERROR 1
ERROR 2
ERROR 2
ERROR 2
ERROR 2
ERROR 2
Password and Confirm password must match.
If you have an ACS member number, please enter it here so we can link this account to your membership. (optional)
ERROR 2
ACS values your privacy. By submitting your information, you are gaining access to C&EN and subscribing to our weekly newsletter. We use the information you provide to make your reading experience better, and we will never sell your data to third party members.
Business
Have antisense oligonucleotides hit their stride?
After 40 years, the drug class looks poised to finally fulfill the promise of rapid development and enter a new era of personalized medicine
by Ryan Cross
November 2, 2018
| A version of this story appeared in
Volume 96, Issue 45

Credit: Ionis Pharmaceuticals
Two antisense oligonucleotides with commonly used modifications that increase the molecule’s stability and affinity for its target. One (below) also has a N-acetylgalactosamine molecule (in gray) conjugated to increase delivery to the hepatocytes of the liver.
Almost two years ago, Julia Vitarello and Alek Makovec learned that their six-year-old daughter, Mila Makovec, has a rare, inherited neurodegenerative condition called Batten disease. The diagnosis explained why the previously healthy girl’s speech, vision, and ability to walk had suddenly started deteriorating. Her brain cells were dying, and doctors delivered the crushing news that the disease, which had no cure, was fatal.
Then Timothy Yu, a researcher at Boston Children’s Hospital, learned about Mila’s story. Her parents were trying to get her genome sequenced to better understand the root cause of her disease. Yu offered to help. When he scanned Mila’s genome, he spotted the genetic glitch that was spurring progressive cell death in her brain: an unwanted stretch of viruslike DNA called a retrotransposon was impeding the function of a critical housekeeping gene in Mila’s cells.
What he did next has made heads spin in the biotech world. In less than a year, Yu developed a drug to help Mila’s cells overlook the disruptive retrotransposon. He first tested it in patient-derived cells in the lab and then sought special permission from the U.S. Food & Drug Administration to give the experimental therapy to Mila. She received her first dose of what Yu named milasen, in her honor, in January.
Mila isn’t cured, but her disease has slowed. This story, which Yu shared at the American Society of Human Genetics meeting in San Diego last month, has rekindled conversations about the promises and perils of personalized medicine.
It has also fueled new interest in the particular kind of drug that Yu made. Called an antisense oligonucleotide, such therapies are particularly well suited to personalization because they are made of nucleotides—the building blocks of our genetic code. Scientists can easily arrange those building blocks in an antisense, or opposing, fashion to repress any sequence of RNA and prevent the production of harmful proteins. In Mila’s case, Yu used an oligonucleotide to repress only the bad part of an RNA containing the retrotransposon, enabling her cells to produce functional proteins that she was lacking before.
The concept isn’t new. In 1978, two scientists demonstrated that a single-stranded antisense oligonucleotide could inhibit viral replication in the lab. In the four decades since, antisense hype has waxed and waned. Double-stranded antisense drugs called small interfering RNAs (siRNAs), which work in a conceptually similar but mechanistically distinct way, also emerged.
Drug companies have tried using oligonucleotides and siRNAs to treat a bevy of infections, chronic conditions, rare genetic diseases, and even cancer. Many of these late-stage clinical trials failed to yield an FDA-approved drug—sometimes because the treatments were ineffective and other times because sporadic, unexplained toxicity derailed a promising program. Only two antisense drugs were approved before 2016, and both were voluntarily pulled from the market because of poor sales.

Credit: Mike Moazami
Interest in treating brain diseases with oligonucleotides is exploding. Here, Jonathan Watts's lab at the University of Massachusetts Medical School tested delivery of an oligonucleotide (green) into the cerebellum of a mouse model of Huntington’s disease.
“It’s been an idea that everyone has felt is about to go big for all those years,” says Jonathan Watts, who develops oligonucleotide therapeutics at the University of Massachusetts Medical School. “It’s been way more complicated than we thought.”
Despite the setbacks, many scientists and investors think things are looking up for the field. FDA has approved three high-profile antisense oligonucleotide drugs since 2016. In response, biotech start-ups are raising hundreds of millions of dollars to create the next generation of treatments. This year, big pharma firms have doled out more than $1 billion to the longtime oligonucleotide specialist Ionis Pharmaceuticals to create new drugs for cancer, cardiometabolic conditions, and neurodegenerative diseases. And academics like Watts are moving their own drugs from lab bench to clinic with breathtaking speed.
Scientists may disagree over whether personalized oligonucleotides will ever be practical, but Mila’s story proves that it is possible. And seemingly everyone in the field agrees that the potential for rapid drug development is what makes the technology so powerful.
Antisense advances
Not long ago, many scientists would have considered developing an antisense drug to treat Mila’s disease, which manifests in the brain, a losing proposition. It’s hard enough to sneak a small molecule across the brain’s protective barriers, let alone something as large as an oligonucleotide. That all changed in 2016 when FDA approved Spinraza, an oligonucleotide developed by Ionis and Biogen to treat the rare childhood neurodegenerative disease spinal muscular atrophy.
Spinraza boosts levels of a critical protein that is deficient in infants with the disease, and it’s saving lives. It requires a direct injection into the spine but fortunately lingers in the connected brain and spinal cord long enough to be effective for several months. “That wasn’t anticipated in the early days,” says Spinraza’s coinventor Adrian Krainer, a scientist at Cold Spring Harbor Laboratory.
Krainer’s collaboration with Ionis began in 2004, and Spinraza was approved 12 years later. The scientific progress the team made laid the foundation for other neurological treatments. Yu was able to make Mila’s drug in under a year. “No one would have tried this before Spinraza was approved,” says David Corey, a nucleic acid chemist at the University of Texas Southwestern Medical Center.
That breakneck pace is unlikely to be regularly repeated, but moving forward, antisense drug development will still be fast compared with development of other drug classes, Corey says. Scientists can screen hundreds of oligonucleotide compounds and pick the best ones in mere months, thanks to their straightforward structures, he explains.
The challenge is not ‘Can we do it?’ but ‘How do we make a business out of it?
Ed Kaye, CEO, Stoke Therapeutics
Oligonucleotides are composed of three main components that chemists can tinker with to improve the drug: the nucleobases that target a complementary RNA sequence, the sugar backbone, and the linkages connecting these sugars, called phosphodiester bonds.
Today, scientists have an established palette of proven modifications that they can mix and match in their oligonucleotides (see “Evolution of Oligonucleotides”). “Once we pick a design we like, all we do is change the sequence. The backbone chemistry and all other elements stay the same,” explains Punit Seth, vice president of medicinal chemistry of Ionis.
Watts and his UMass colleague Anastasia Khvorova have coined a term for why one successful antisense therapy can help spur the creation of so many more: the dianophore. It’s a spin on “pharmacophore,” a term drug designers use to describe how the structure of a small molecule interacts with its target, usually a protein. An oligonucleotide’s pharmacophore, and the RNA target it binds, is determined simply by a sequence of nucleotides. Changing the sequence changes the target. But chemists can also modify the backbone of an oligonucleotide to alter its pharmacokinetics—the drug’s distribution and stability in the body. Watts and Khvorova call these pharmacokinetic properties the dianophore.
For small-molecule drugs, tweaking the structure changes both the pharmacophore and dianophore, but not always in the desired way—a modification to enhance a drug’s distribution, for example, might loosen its hold on its target. For oligonucleotides, Watts and Khvorova posit that the pharmacophore and dianophore are largely independent. And since changing the sequence, or pharmacophore, of an oligonucleotide is easy, when scientists land on a really great dianophore, “that’s when you see these really rapid explosions in the field,” Watts says.
That explosion is underway with oligonucleotides targeting diseases in the spine and brain, thanks to Spinraza. It’s also ongoing with antisense drugs designed to treat liver diseases, because of a molecule called N-acetylgalactosamine, or GalNAc, that selectively shuttles oligonucleotides into hepatocytes, a kind of liver cell.
In other cases, that burst of activity has gotten ahead of the science. After FDA approved Exondys 51, Sarepta Therapeutics’ drug for Duchenne muscular dystrophy, in 2016, many others pursued oligonucleotide therapy for muscle diseases. But clinical data for Sarepta’s drug were thin, and many people doubt its effectiveness. Scientists think new chemical strategies will be needed to help get higher levels of oligonucleotides into muscle cells to treat muscular diseases.
Evolution of oligonucleotides
Over the decades, chemists have tinkered with oligonucleotides and made dozens of modifications to improve the molecules’ stability and potency. Many of them never caught on. Here are the creations that stuck and are being used or tested in humans now.
Phosphorothioate backbone: The natural phosphodiester bond that links the sugars in DNA and RNA is quickly cleaved by enzymes called nucleases. Replacing an oxygen with a sulfur creates a nuclease-resistant phosphorothioate bond, but it also introduces a chiral center. Interest in controlling the chirality of each linkage has increased recently.
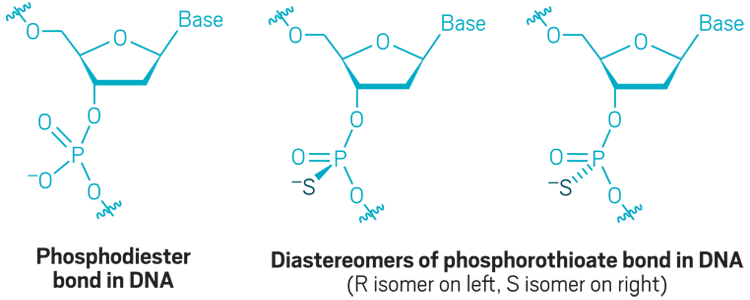
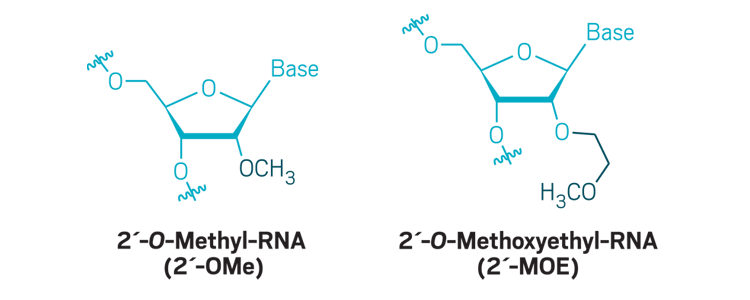
Sugar modifications: Modifications to the 2´-carbon of ribose are commonly used to boost nuclease resistance and increase the binding of the antisense oligonucleotide to its target.
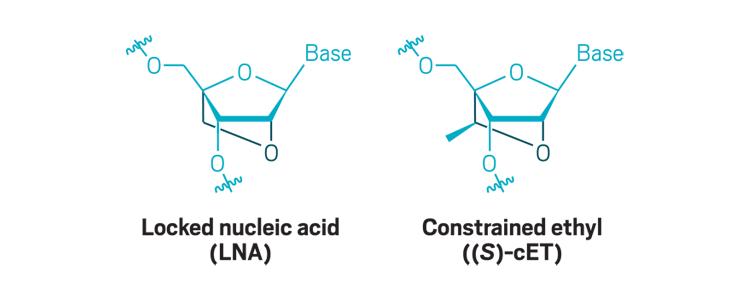
Bridged nucleic acids: Creating a bridge between the 2´-oxygen and 4´-carbon of ribose dramatically increases an antisense oligonucleotide’s binding affinity. These modifications are usually added to only portions of the molecule.
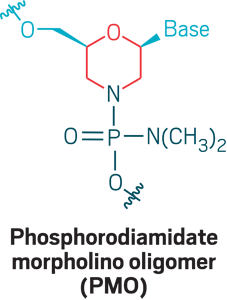
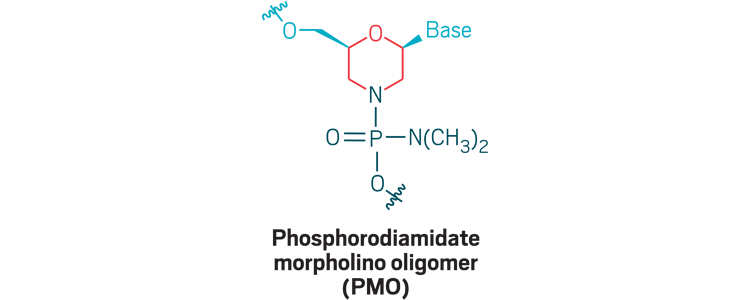
Morpholinos: Phosphorodiamidate morpholino oligomers (PMOs) are oligonucleotide analogs that use a morpholino ring (red) and phosphorodiamidate (dark blue) linkage to create a neutrally charged strand that can bind RNA for exon skipping, wherein cells skip over a stretch of genetic code. PMOs don’t cross cell membranes well, so new versions incorporate positive charges or append positively charged peptides to facilitate delivery.
Oligo engineering
At the end of September, as the Oligonucleotide Therapeutics Society annual meeting kicked off in Seattle, the question on everyone’s minds was simple: What will catalyze the next big explosion in the field?
The answer is complicated. All the approved drugs use old chemistry: The oligonucleotide analog used to make Exondys 51 was invented in the 1980s, as were the phosphorothioate linkages used widely in earlier treatments; the 2’-O-methoxyethyl modificationused in Spinraza was invented in 1996; the locked nucleic acids Roche uses were first made in 2002, and Ionis made a similar modification called a constrained ethyl in 2009. Since then, scientists have been slow to adopt new chemistries in preclinical or clinical studies.
Most of the effort recently has gone into using these existing chemistries in novel combinations to make therapies safer and more potent. The most common side effect of oligonucleotide therapy is thrombocytopenia, a dangerously low platelet count that increases the risk of bleeding. It’s a problem for some, but not all, oligonucleotides containing negatively charged phosphorothioates, including several high-profile drugs that FDA rejected. One that got the green light from regulators this October, an Ionis and Akcea Therapeutics rare disease drug called Tegsedi, carries a warning about the condition.
Advertisement
At the Oligonucleotide Therapeutics Society meeting, Ionis’s Seth shared highlights of a project to replace phosphorothioates with neutrally charged modifications along certain sites of the oligonucleotide backbone to improve safety while retaining efficacy.
Chandra Vargeese, senior vice president of drug discovery at Wave Life Sciences, sees another problem with phosphorothioates. “All good things come with baggage,” she says, explaining that every sulfur in the linkage creates a chiral center, resulting in two versions of the molecule called diastereomers with different spatial arrangements. That means a typical oligonucleotide with 20 bases and 19 linkages will actually be a mixture of 524,288 diastereomers.
Companies had acknowledged, but mostly ignored, that fact over the decades, until Wave stepped in to explore the therapeutic benefits of stereopure oligonucleotides—in which the chirality of each phosphorothioate is precisely controlled. The company is currently running several closely watched clinical trials. Roche also quietly started its own stereopure oligonucleotide program.
At the Oligonucleotide Therapeutics Society meeting, Vargeese presented data suggesting that Wave’s stereopure molecules worked better than standard random mixtures in a mouse model of muscular dystrophy. But many people at the meeting said such results need to be taken with a grain of salt. “There is always a new version, and the version n plus one is often pointed to as the panacea,” says Paul Burke, an oligonucleotide consultant who formerly worked at Pfizer and Merck & Co. Companies talk about how new chemistries or stereopurity can increase efficacy and decrease toxicity, “but they don’t get into the mechanisms,” he says.
Those mechanisms may become even harder for outsiders to decipher as the industry begins taking a big-data approach to oligonucleotide design and development. For instance, Morten Lindow, a director of RNA therapeutics research at Roche, says the company has developed algorithms to predict cell and liver toxicity from a nucleotide sequence. The big pharma firm also screens hundreds of oligonucleotides targeting different regions of the same RNA to see which works best. “This means that to a large extent the traditional role of a medicinal chemist is carried out by bioinformaticians,” he explains.
Hope and hype
The oligonucleotide field is full of fresh momentum, and cash. And although many companies have high hopes for replicating Spinraza’s success, researchers who have ridden out the field’s many setbacks are cautious about the future.
Still, the excitement in the field is palpable. When the news about Yu’s personalized treatment for Mila broke, it spread like wildfire throughout the antisense oligonucleotide community. “The milasen story is unbelievably impressive, and it generates a lot of dreams,” says Marc Lemaitre, an antisense consultant. “I don’t see any other type of class of drug that can be developed so quickly. That is the power of oligonucleotides.”
There is always a new version, and the version n plus one is often pointed to as the panacea.
Paul Burke, independent oligonucleotide consultant
Paula Evans, CEO of a new oligonucleotide-focused start-up called GeneTx Biotherapeutics, expects more academic labs to follow in Yu’s footsteps. In fact, she is working with Yu and Texas A&M University scientist Scott Dindot to create therapies for the neurological condition Angelman syndrome.
But Evans does not expect that GeneTx’s Angelman program, or others like it, will progress at the breakneck speed seen with milasen. Regulatory authorities were more flexible with their requirements because Mila likely would have died before there was even time to make an animal model. When a disease is not so rapidly degenerative or fatal, regulators have higher standards for safety. And the rarity of Mila’s mutation was also a factor. Whereas her drug might not help anyone else, a disease like Angelman affects enough people that companies can run clinical trials to understand the risk and efficacy of a treatment. In those cases, “we have to go through a much more rigorous process, because we can,” she says.
That leads to another concern: cost. The total price of Mila’s experimental therapy hasn’t been disclosed, and Boston Children’s Hospital did not respond to repeated interview requests with C&EN. But Mila’s parents raised $1.5 million in online donations, and experts suggest milasen’s development likely cost double that.
It’s hard to imagine how a drug company could ever recoup the investment made in such individualized therapies, especially when safety testing in monkeys can easily cost more than $1 million, Lemaitre says. The situation has left companies to figure out how to turn a profit with complex treatments for small patient populations. “The challenge is not ‘Can we do it?’ but ‘How do we make a business out of it?’ ” says Ed Kaye, who was CEO of Sarepta Therapeutics during the development of Exondys and now heads a new oligonucleotide-focused start-up, Stoke Therapeutics.
Those problems make oligonucleotide therapies ripe for development by university scientists studying rare diseases. “Academic labs like mine can push into this space in ways that we might not be able to for traditional small-molecule drug discovery,” Watts says.
It makes for an exciting time in a field that has managed to survive many disappointments. In the next few years, everyone will be closely watching the key clinical trials—they will be the true test as to whether the promise of rapid antisense drug development has finally arrived.
At the minimum, people are hopeful that the time it takes to go from idea to human tests will be shorter. “The risk of developing these drugs is going to be lower, and the speed will be faster,” UT Southwestern’s Corey says. “This could be a great way to make personalized medicines quickly. But you can’t check your skepticism at the door.”
Chemical & Engineering News
ISSN 0009-2347
Copyright © 2024 American Chemical Society
You might also like...



The power is now in your (nitrile gloved) hands
Sign up for a free account to get more articles. Or choose the ACS option that’s right for you.
Already have an ACS ID? Log in
Join the conversation
Contact the reporter
Submit a Letter to the Editor for publication
Engage with us on Twitter