Advertisement
When the first human genome was sequenced, researchers finally got a glimpse of the true dimensions of our proteome—the tens of thousands of proteins responsible for sickness and health. Ever since, scientists have explored the morass for drivers of diseases, generating a long wish list of proteins they’d like to control.
In brief
Targeted protein degradation, a way of sending bad-behaving proteins to the cellular trash compactor, is a hot new area in drug discovery. In just two years, multiple companies have bought into the idea that bifunctional small molecules can open up a broad swath of the human proteome. But much work remains to translate what was until recently an academic endeavor into a commercial opportunity. Read on to find out how biotech and big pharma firms are trying to take protein degraders from the lab to the marketplace.
But even as they add to the list, drug hunters live with a maddening reality: So many of its entries are out of reach. Conventional small-molecule and antibody drugs can access only about 20% of the proteins we make.
Advertisement
So when a new technology comes along promising to tap into the other roughly 80%, everyone pays attention. Over the years, approaches such as RNA-based silencing and gene editing have raked in billions of dollars in financing for their potential to turn off the activity of previously unreachable proteins.
But new modalities like these come with growing pains—delivery problems, potency issues, safety questions—that make the arc from idea to marketed treatment excruciatingly long. What if instead you could reimagine an old workhorse—the small-molecule drug—into a new kind of medicine?
Researchers developing targeted protein degraders aim to do just that. Conventional small molecules block the activity of a protein. In contrast, these bifunctional small molecules eliminate the protein altogether by routing it to the proteasome, the cell’s trash compactor.
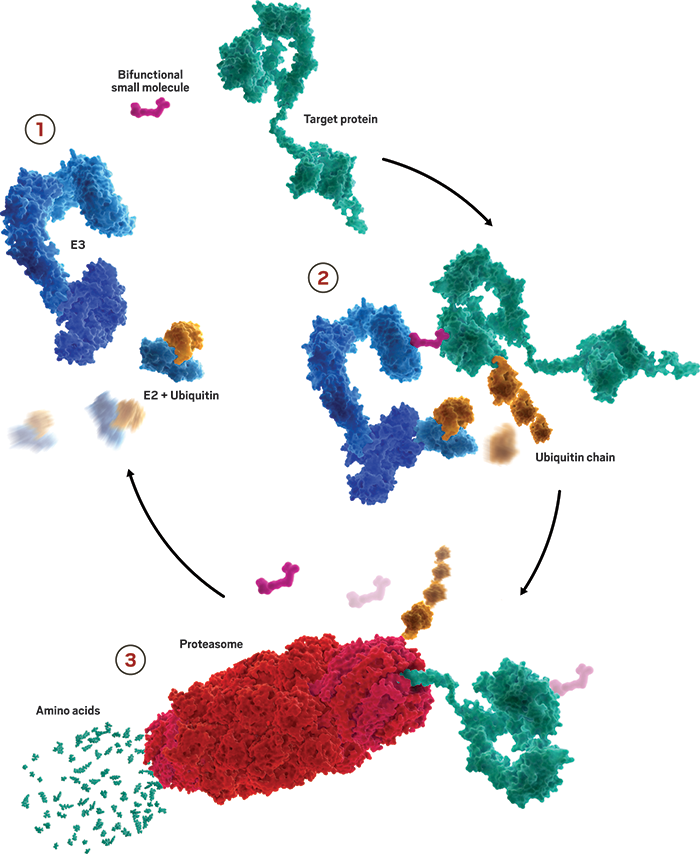
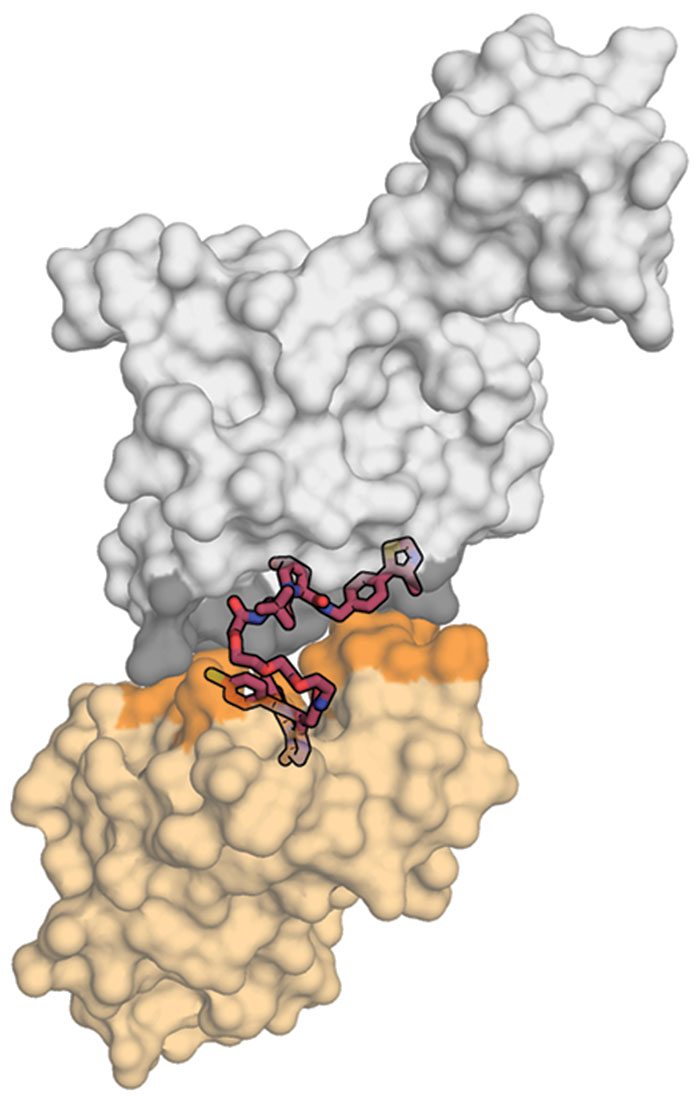
Our cells are always working to maintain just the right levels of proteins—making and disposing tens of thousands of them at any given time. A key player in managing that balance is a small protein called ubiquitin. When tacked onto exhausted proteins, it routes them to the proteasome for disposal.
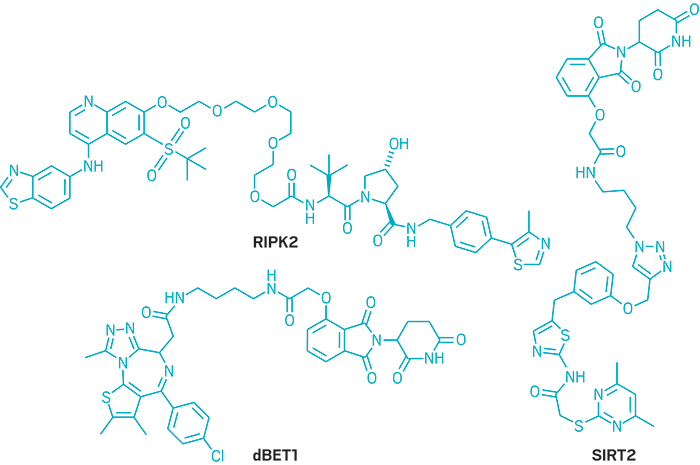
Researchers developing protein degraders want to break into that ubiquitin-proteasome machinery to change the destiny of disease-causing proteins. To do that, they design small molecules with two active ends: one that binds to the protein of interest and the other that binds to a protein called E3 ubiquitin ligase. These bifunctional molecules force a handshake between the ubiquitin and the protein, sending it to the trash. The job done, they move on to repeat the process, quickly depleting levels of the unwanted protein.
It sounds devilishly simple. But protein degraders break all the rules about how a small molecule looks and behaves. To accommodate both functional parts, the molecules tend to be bulky on the ends and floppy in the middle. They also weigh more than is typically considered acceptable for a drug.
Unlike conventional drugs, these molecules act kind of like an enzyme or catalyst, with each one sending multiple target proteins to the trash. And for reasons still being explored, they upend conventional wisdom about drug potency and selectivity.
Because of those quirks, protein degraders might easily have been relegated to the annals of academic oddities. Indeed, not even five years ago, “people were queuing up to tell us the reasons why this wouldn’t work,” says Ian Churcher, vice president of drug discovery at BenevolentAI. Churcher previously led GlaxoSmithKline’s protein degradation unit, which was an early adopter when it launched in 2012. “To many medicinal chemists, these structures were truly strange.”
But thanks to a flurry of academic work proving, in cells at least, that targeted degradation is a powerful way to silence errant proteins, industry has taken notice. Since 2016, multiple protein-degradation-focused biotech firms have emerged with ample funding. Big pharma companies have launched internal efforts and forged partnerships to explore the modality.
Momentum is building, but drug company scientists still need to confront a few critical questions. The biggest—whether this new modality will be safe and effective in humans—could be answered soon. Arvinas, one of the first protein-degrader companies to emerge, plans to put two cancer drug candidates into clinical studies this year.
Researchers also need to push beyond the status quo. The molecules published so far were discovered empirically and act against well-established proteins. If companies want to access the other 80% of the proteome, their medicinal chemists will need to forge a new set of rules for how to rationally build drugs that consistently degrade proteins.
Research executives seem confident that the remaining challenges boil down to elbow grease. As Jay Bradner, president of the Novartis Institutes for BioMedical Research, puts it, “Our field is really good at reducing concepts to practice.”