Advertisement
Grab your lab coat. Let's get started
Welcome!
Welcome!
Create an account below to get 6 C&EN articles per month, receive newsletters and more - all free.
It seems this is your first time logging in online. Please enter the following information to continue.
As an ACS member you automatically get access to this site. All we need is few more details to create your reading experience.
Not you? Sign in with a different account.
Not you? Sign in with a different account.
ERROR 1
ERROR 1
ERROR 2
ERROR 2
ERROR 2
ERROR 2
ERROR 2
Password and Confirm password must match.
If you have an ACS member number, please enter it here so we can link this account to your membership. (optional)
ERROR 2
ACS values your privacy. By submitting your information, you are gaining access to C&EN and subscribing to our weekly newsletter. We use the information you provide to make your reading experience better, and we will never sell your data to third party members.
Look back at 2007
C&EN steps back in time to look at research advances from a decade ago
December 18, 2017 | Volume 95 Issue 49
G protein-coupled receptor structures aided drug design
A decade after a key structural analysis, scientists have a better understanding of the pharmaceutically important family of receptor proteins
By Stu Borman
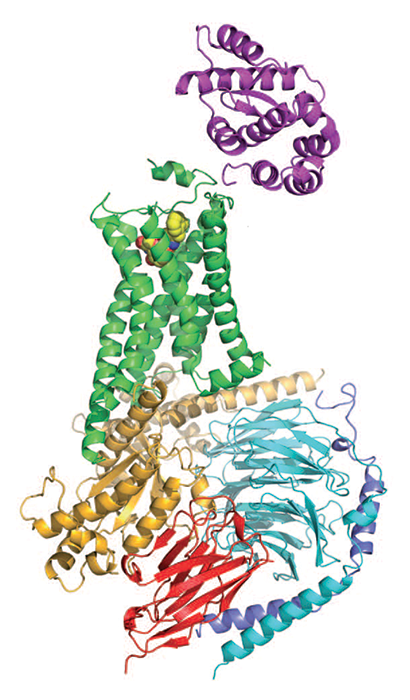
G protein-coupled receptors, or GPCRs, are the largest family of human proteins, with over 800 members. They reside within cell membranes, receiving molecular signals from outside the cell and translating them into signals that influence cellular pathways inside the cell.
In 2000 researchers obtained the first GPCR crystal structure, and a decade ago they got the second one. The analysis of GPCR structures began a new chapter in drug discovery, enabling researchers to better design GPCR-targeted medicines.
When a signaling molecule, such as a hormone, binds to the part of a GPCR that protrudes from the cell, the receptor undergoes a conformational change that activates a G protein (guanine nucleotide-binding protein) bound to the part of the GPCR that's inside the cell. The activated G protein then modulates the activity of other proteins and signaling pathways.
GPCRs are targets of many drugs designed to influence those pathways, including treatments for ulcers, migraines, cancer, and stroke. According to GPCRdb, a web-based storehouse of GPCR data and design tools, nearly 500 drugs—about one-third of all FDA-approved medications—act on over 100 GPCR targets. More than 300 additional GPCR-targeted drug candidates are being clinically tested (Nat. Rev. Drug Discovery 2017, DOI: 10.1038/nrd.2017.178).
Researchers early on realized that obtaining the X-ray crystal structures of GPCRs could aid drug design. But GPCRs are tough nuts to crack structurally. Their extended, floppy conformations help them do their jobs but make them hard to isolate from cell membranes in functional and stable forms. Also, scientists often must express the proteins in genetically modified bacteria to produce quantities sufficient for crystallization.
Researchers obtained the first GPCR crystal structure, of rhodopsin, by isolating the protein from cow retinas, crystallizing it, and working in a cold, dark room for two years to get the light-sensitive crystals to remain stable (Science 2000, DOI: 10.1126/science.289.5480.739). It took another seven years for scientists to report structures of a second GPCR, human β2 adrenergic receptor (β2AR). Brian K. Kobilka of Stanford University School of Medicine, Raymond C. Stevens of Scripps Research Institute California, and coworkers analyzed a fusion protein of β2AR with a stabilizing enzyme (Science 2007, DOI: 10.1126/science.1150577). And Kobilka and coworkers analyzed β2AR after stabilizing it with an antibody fragment (Nature 2007, DOI: 10.1038/nature06325).
Chris Tate of MRC Laboratory of Molecular Biology and coworkers had earlier used point mutations to stabilize GPCRs without disrupting the proteins’ conformations or functions. The researchers, including Gebhard Schertler, obtained a GPCR structure with the method in 2008. Heptares Therapeutics later obtained rights to that method. Now called StaR, this approach has helped researchers obtain a number of GPCR structures.
Despite these successes, no structures showed a GPCR interacting with a G protein, making it difficult to understand exactly how the receptors activated the proteins. But in 2011, Kobilka, Roger K. Sunahara of the University of California, San Diego, and coworkers finally captured that interaction by using an enzyme, an antibody, detergents, and lipids to stabilize β2AR bound to its G protein (Nature 2011, DOI: 10.1038/nature10361). The study helped researchers begin to nail down the mechanism of G protein activation, in which guanosine diphosphate dissociates from a G protein, guanosine triphosphate replaces it, and two parts of the G protein dissociate to regulate other proteins.
Progress in the field was recognized the next year when Robert J. Lefkowitz of Duke University and Kobilka took home the 2012 Nobel Prize in Chemistry for their work on GPCRs. Lefkowitz’s group, beginning in 1968, isolated some of the receptors and studied how they work. While Kobilka was a postdoc in Lefkowitz’s lab, the pair determined that such receptors belonged to a large family, later named GPCRs.
Researchers have now obtained about 50 high-resolution GPCR crystal structures, most under the auspices of the U.S. National Institutes of Health Protein Structure Initiative’s GPCR Network and a successor group, the GPCR Consortium.
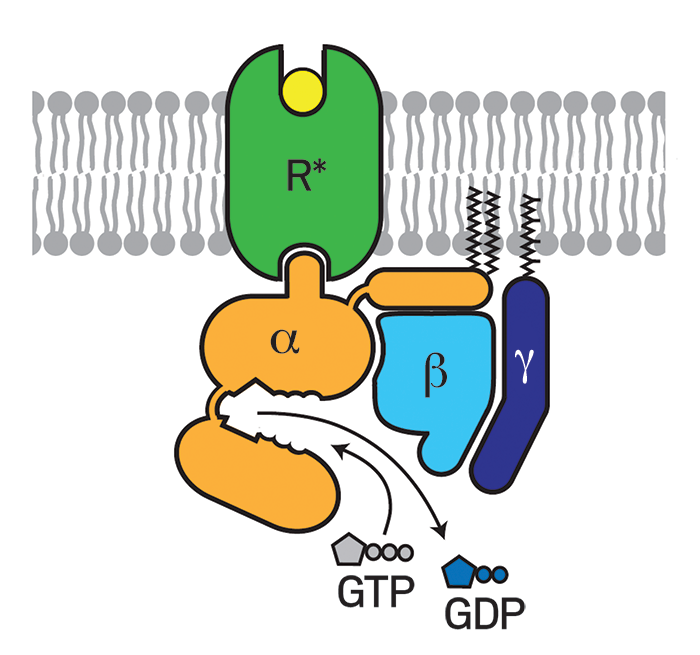
The benefits of GPCR structural information continue to grow. For example, last year researchers computationally docked a virtual library of druglike compounds with a GPCR structure determined by Kobilka’s lab to find PZM21, an analgesic that reduces pain in mice longer than morphine does, and with fewer side effects. The GPCR company Receptos used structure-based design to identify RPC1063 (ozanimod), a GPCR-targeted drug candidate that Celgene is testing in clinical trials for multiple sclerosis, ulcerative colitis, and Crohn’s disease. Crystal structures also helped Heptares design the GPCR-based drug candidate AZD4635, which AstraZeneca is testing to treat cancer.
“The past 10 years have seen dramatic improvements in our ability to solve X-ray structures of GPCRs and utilize these for drug design,” says Fiona Marshall, director and chief scientific officer of Heptares. GPCRdb director David E. Gloriam of the University of Copenhagen adds that genomic, structural, and functional GPCR information will increase even more rapidly going forward, enabling researchers “to better understand and target disease from the molecular to the physiological scale.”
Covalent organic frameworks debuted and multiplied quickly
The variety of porous, metal-free crystals and their applications has grown, but no signs yet of commercialization
By Mitch Jacoby
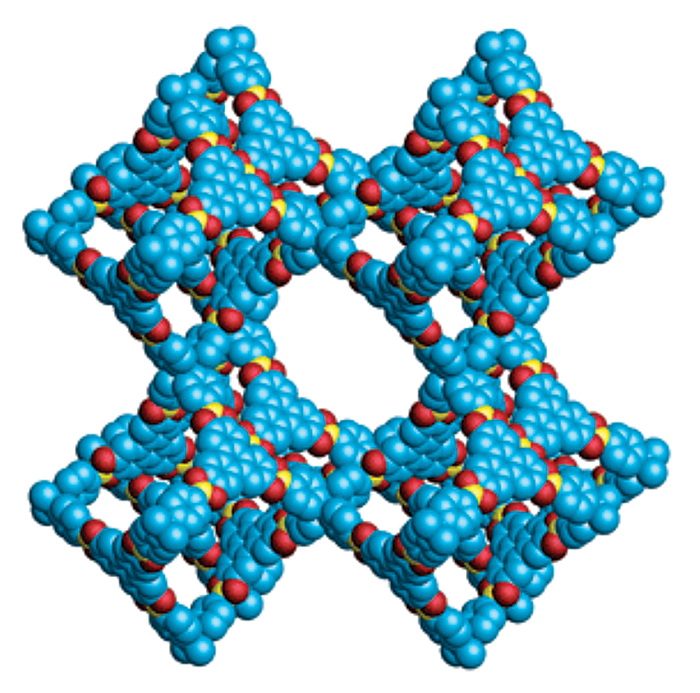
In a 1993 essay in Scientific American, chemistry Nobel laureate Roald Hoffmann of Cornell University reflected on the ability of chemists to systematically construct highly complex molecules. “Organic chemists are masterful at exercising control in zero dimensions,” he noted, referring to the creation of single, small molecules. Polymer chemists, he said, extend that control to one dimension by building molecular chains. “But in two or three dimensions, it’s a synthetic wasteland,” Hoffmann wrote, suggesting that chemists of the time were not yet skilled at making organic structures that extended “infinitely” in space.
That situation started to change in 2005 when Omar M. Yaghi and his colleagues synthesized two-dimensional, porous, crystalline networks composed entirely of light elements, such as carbon, boron, and oxygen, held together by strong covalent bonds. In 2007, Yaghi, now at the University of California, Berkeley, and coworkers made the first 3-D versions of these materials, dubbed covalent organic frameworks, or COFs (Science 2007, DOI: 10.1126/science.1139915). The COFs exhibited intriguing properties, including very low density, exceptionally high surface area, and high thermal stability—all features that suggested these materials may be useful for storing and separating gases.
By the time 3-D COFs came on the scene, Yaghi and other researchers had already spent more than a decade synthesizing a related and now better-known class of porous, crystalline materials called metal-organic frameworks, or MOFs. Unlike COFs, MOFs contain metal ions or clusters of metal ions connected by organic linkers. COFs, on the other hand, are constructed entirely from covalently bonded organic building blocks and so have none of the coordination bonding characteristics of metal complexes.
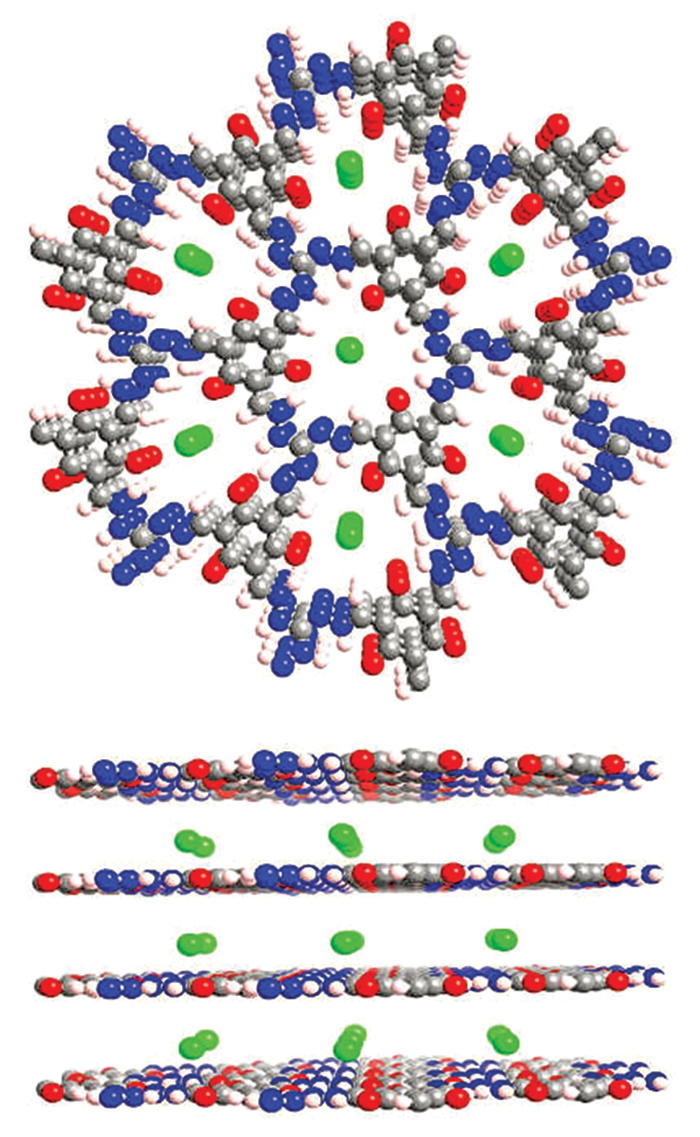
As a rule of thumb, the all-organic nature of COFs renders them more stable than MOFs against degradation by water, acids, and bases. According to William R. Dichtel, a COF specialist at Northwestern University, the fully organic quality of COFs also means researchers can draw from the rich assortment of tools in the organic chemistry toolbox to precisely tailor the size and shape of COF pores and carefully position a wide range of functional groups inside the pores. The ability to fine-tune the structure and composition of COFs enables scientists to customize the crystals to suit intended applications.
In the decade since COFs made their debut, dozens of research groups have climbed aboard the porous framework train and have published hundreds of papers detailing a wide variety of COFs and COF applications. However, none of the researchers C&EN contacted for this story is aware of plans to commercialize these materials.
“It’s just too early,” Dichtel says. “We haven’t come up with a killer application that the market will pay for.” Dichtel points out that MOF commercialization began just recently, and those materials had a roughly 15-year head start over COFs.
Nonetheless, COF development marches onward. In addition to using the materials to store and separate gases such as carbon dioxide, methane, and hydrogen, researchers have shown that these materials can serve as water-purification membranes to remove organic pollutants, salts, and metal ions. COFs have been used as charge conductors and as energy storage materials for batteries and capacitors. The porous compounds have also been used as catalysts to facilitate condensation reactions in organic synthesis and electrochemical reduction of CO2 to CO, which is important for industrial production of hydrocarbons. And the materials have even been used to make membranes with antimicrobial properties.
Taking COFs in a different direction, Yaghi and coworkers reported last year that the materials can be used as “molecular threads” to form interwoven structures with reversible elasticity. The woven materials, which bend and stretch without breaking bonds, eventually may lead to new types of flexible thin films and electronic devices (Science 2016, DOI: 10.1126/science.aad4011).
“We’re always interested in applications,” Yaghi says. “But as chemists, first and foremost, we think about new kinds of molecules, bonds, and reactions.” As chemists continue to fine-tune the properties of COFs, applications will surely follow, he adds.
Reversing Rett syndrome in mice gave hope for treatments
Experiment demonstrated that fixing the molecular basis for the disease could restore lost function
By Michael Torrice
Ten years ago, Adrian Bird of the University of Edinburgh published a paper in Science that changed how scientists think about Rett syndrome, a rare genetic brain disorder with some autismlike symptoms that affects mostly girls.
Through genetic engineering, Bird and his colleagues turned off the gene in mice that stops working in Rett. As expected, with the gene inactive, these animals developed Rett-like symptoms, including abnormal breathing. But when the researchers reactivated the gene, the mice basically returned to normal (Science 2007, DOI: 10.1126/science.1138389). The experiment suggested that if scientists could somehow find a way to repair the molecular basis of Rett in people, they could treat, and possibly cure, the disease.
The 2007 paper “was a paradigm shift,” says David M. Katz of Case Western Reserve University School of Medicine. “If you spoke to any clinician before then and said that this disorder might be reversible, they’d have looked at you like you were crazy.”
Scientists and clinicians once viewed Rett as they do Alzheimer’s or Parkinson’s—as diseases that cause irreversible brain damage, says Randall Carpenter, the chief scientific officer for the Rett Syndrome Research Trust (RSRT), a patient advocacy group that funds research on Rett therapies.
Besides motivating Rett researchers, Bird’s experiment “also gave hope to families that, even though their children had already regressed, we may someday be able to treat them,” he adds. One parent who found inspiration from the 2007 study was Monica Coenraads, who started RSRT in 2008 and is the organization’s executive director.
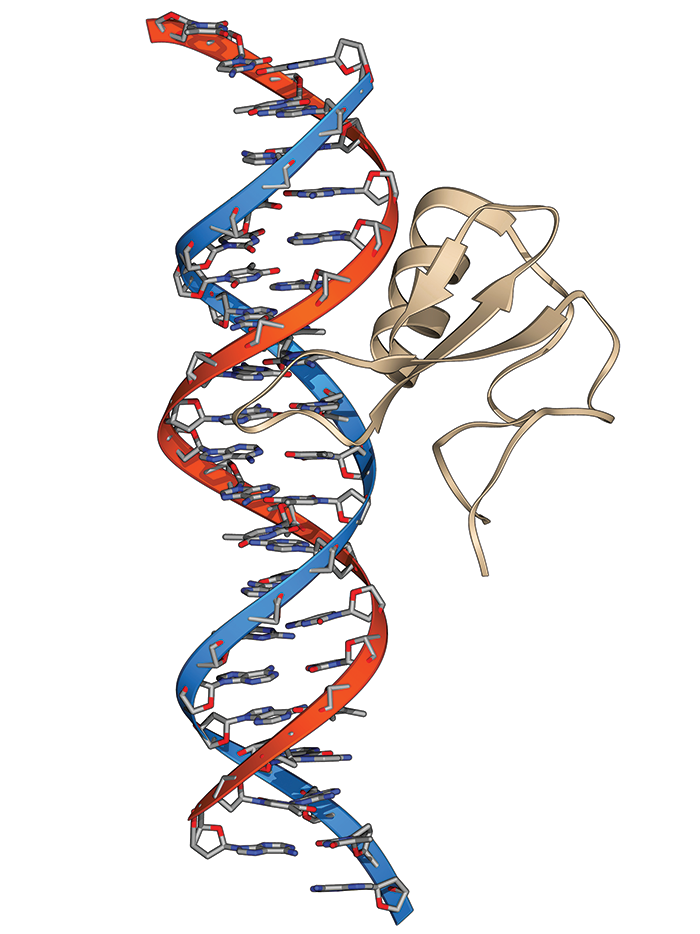
Most of RSRT’s efforts today focus on therapies targeting the gene behind Rett—MECP2. It codes formethyl-CpG-binding protein 2 (MeCP2), and mutations to the gene leave the protein inactive, leading to abnormal expression of myriad genes in the brain.
Children with mutations in MECP2, which sits on the X chromosome, develop normally for the first year and then start to regress. They develop symptoms such as the inability to speak, difficulty with walking, and loss of control of their hands.
The therapeutic strategy to reverse Rett in people that has received the most attention in the past decade is gene therapy—delivering a healthy copy of the MECP2 gene to brain cells. In June of this year, AveXis announced plans to develop a gene therapy for Rett based on a viral delivery system the company previously used in a Phase I clinical trial for spinal muscular atrophy.
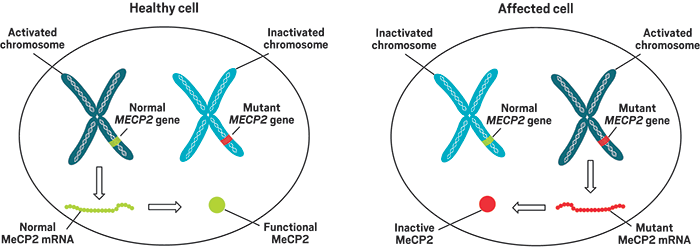
Preclinical research in animals over the past decade suggested that too much expression of the MeCP2 protein also impairs brain function, Carpenter says. To produce a Rett gene therapy safe enough for testing in people, scientists have designed payloads that carry not only healthy copies of MECP2 but also genetic elements that regulate expression of that gene.
Other researchers are working on therapies that target pathways affected by the loss of the protein MeCP2. Such treatments wouldn’t cure Rett, but they could help people improve their quality of life by addressing symptoms of the disease, says Colleen Niswender of the Vanderbilt Center for Neuroscience Drug Discovery.
Loss of MeCP2 leads to many molecular and electrical signaling problems in the brain. These downstream treatments attempt to restore that signaling. For example, Katz and others have studied how ketamine can turn up the volume on certain electrical circuits that have become quiet in Rett brains. He’s currently involved in a Phase II clinical trial of low doses of ketamine in people with Rett.
A decade after the 2007 Rett study, scientists studying rare genetic disorders still talk about the experiment, says Stuart Cobb of the University of Edinburgh, who was a coauthor on the paper. “Because of that paper,” he says, “Rett is held as an exemplar of rare disorders and how you can test the reversibility of them to show that there is merit in going forward for cures.”
Chemistry then and now: 2007 versus 2017
C&EN compares snapshots of the chemistry scene today and a decade ago

